Вся информация, размещенная в данном веб-сайте, предназначена исключительно для специалистов здравоохранения/медицинских работников.
Пожалуйста, подтвердите, что Вы являетесь медицинским или фармацевтическим работником.
Каждая таблетка дозировкой 850 мг + 10 мг содержит:
Действующие вещества:
метформина гидрохлорид – 850,0 мг; сибутрамина гидрохлорида моногидрат – 10,0 мг;
Вспомогательные вещества:
целлюлоза микрокристаллическая, кроскармеллоза натрия, повидон К-25, магния стеарат;
Вспомогательные вещества пленочной оболочки:
Готовая система для покрытия Опадрай II 85F30656 голубой (поливиниловый спирт, макрогол, титана диоксид, тальк, краситель бриллиантовый голубой с алюминиевым лаком, краситель индигокармин с алюминиевым лаком, краситель железа оксид желтый).
Каждая таблетка дозировкой 850 мг + 15 мг содержит:
Действующие вещества:
метформина гидрохлорид – 850,0 мг; сибутрамина гидрохлорида моногидрат – 15,0 мг;
Вспомогательные вещества:
целлюлоза микрокристаллическая, кроскармеллоза натрия, повидон К-25, магния стеарат;
Вспомогательные вещества пленочной оболочки:
Готовая система для покрытия Опадрай II 85F48105 белый (поливиниловый спирт, макрогол, тальк, титана диоксид).
Дозировка 850 мг + 10 мг
Таблетки овальные двояковыпуклые с разделительной риской с одной стороны, покрытые пленочной оболочкой голубого цвета. На поперечном разрезе ядро почти белого цвета.
Дозировка 850 мг + 15 мг
Таблетки овальные двояковыпуклые с разделительной риской с одной стороны, покрытые пленочной оболочкой белого цвета. На поперечном разрезе ядро почти белого цвета.
Гипогликемическое средство группы бигуанидов для перорального применения. Средство для лечения ожирения.
Фармакодинамика
Метформин
Пероральный гипогликемический препарат из группы бигуанидов, снижает гипергликемию, не приводя к развитию гипогликемии. В отличие от производных сульфонилмочевины не стимулирует секрецию инсулина и не вызывает гипогликемического эффекта у здоровых людей. Повышает чувствительность периферических рецепторов к инсулину и утилизацию глюкозы клетками. Тормозит глюконеогенез в печени. Задерживает всасывание углеводов в кишечнике и стимулирует выработку ГПП-1 (физиологического регулятора аппетита). Метформин стимулирует синтез гликогена, воздействуя на гликогенсинтазу. Увеличивает транспортную емкость всех типов мембранных переносчиков глюкозы. Кроме того, оказывает благоприятный эффект на метаболизм липидов: снижает концентрацию общего холестерина, липопротеинов низкой плотности и триглицеридов.
На фоне приема метформина масса тела пациента либо остается стабильной, либо умеренно снижается.
Сибутрамин
Является пролекарством и проявляет свое действие in vivo за счет метаболитов (первичных и вторичных аминов), ингибирующих обратный захват моноаминов (серотонина, норадреналина и дофамина). Увеличение содержания в синапсах нейротрансмиттеров повышает активность центральных 5НТ-серотониновых и адренергических рецепторов, способствует физиологической регуляции аппетита за счет увеличения чувства насыщения и снижения потребности в пище, а также увеличению термопродукции (внутреннего расхода энергии). Опосредованно активируя βз-адренорецепторы, сибутрамин воздействует на бурую жировую ткань. Снижение массы тела при приеме сибутрамина сопровождается увеличением концентрации в сыворотке крови липопротеинов высокой плотности (ЛПВП) и снижением концентрации триглицеридов, общего холестерина, липопротеинов низкой плотности (ЛПНП) и мочевой кислоты. Сибутрамин и его метаболиты не влияют на высвобождение моноаминов, не ингибируют моноаминооксидазу (МАО); не обладают сродством к большому числу нейромедиаторных рецепторов, включая серотониновые (5-НТ1, 5-HT1a, 5-HT1b, 5-НТ2с), адренергические (β1, β2, β3, α1, α2), дофаминовые (D1, D2), мускариновые, гистаминовые (H1), бензодиазепиновые и глутаматные NMDA рецепторы.
Одновременное применение метформина и сибутрамина повышает эффективность терапии у пациентов с ожирением. Посредством регуляции аппетита, снижения чувства голода, повышения расхода энергии и регуляции липидного и углеводного обменов Редуксин®Форте уменьшает массу тела человека и восстанавливает метаболическое здоровье.
Клиническая эффективность и безопасность (Результаты клинических исследований)
В клиническом исследовании в группе препарата Редуксин® Форте доля пациентов, достигших клинически значимого снижения веса ≥5% за 3 месяца терапии (доля ранних ответчиков на терапию) превышала 90%. За 6 месяцев терапии 91,67% пациентов в группе Редуксин® Форте достигли снижения массы тела на 10% и более. Снижение массы тела сопровождалось клинически значимым уменьшением окружности талии и улучшением липидного профиля, что доказывает эффективность препарата в отношении снижении риска развития осложнений и отвечает основным целям терапии ожирения.
В ходе исследования не наблюдалось негативного влияния препарата Редуксин® Форте на показатели сердечно-сосудистой системы при его применении у пациентов с ожирением.
Фармакокинетика
Всасывание
После приема препарата внутрь метформин достаточно полно абсорбируется из ЖКТ. При одновременном приеме пищи абсорбция метформина снижается и задерживается. Абсолютная биодоступность составляет 50-60 %. Максимальная концентрация в плазме (Cmax) составляет приблизительно 2 мкг/мл или 15 мкмоль и достигается через 2,5 ч.
Распределение
Метформин быстро распределяется в тканях организма. Практически не связывается с белками плазмы.
Метаболизм
Подвергается метаболизму в незначительной степени.
Выведение
Выводится почками. Клиренс метформина у здоровых людей составляет 400 мл/мин (в 4 раза выше, чем клиренс креатинина (КК)), что свидетельствует об активной канальцевой секреции.
Период полувыведения (T1/2) составляет приблизительно 6,5 ч.
Фармакокинетика в особых клинических случаях
У пациентов с почечной недостаточностью T1/2 возрастает, появляется риск кумуляции метформина в организме.
Сибутрамин
Всасывание
После приема внутрь быстро всасывается из ЖКТ не менее чем на 77 %. При «первичном прохождении» через печень подвергается биотрансформации под влиянием изофермента CYP3A4 с образованием двух активных метаболитов (монодесметилсибутрамин (М1) и дидесметилсибутрамин (М2)). После приема разовой дозы 15 мг максимальная концентрация в крови (Сmах) монодесметилсибутрамина (М1) составляет 4 нг/мл (3,2-4,8 нг/мл), дидесметилсибутрамина (М2) - 6,4 нг/мл (5,6-7,2 нг/мл). Сmах достигается через 1,2 ч (сибутрамин), 3-4 ч (активные метаболиты). Одновременный прием пищи понижает Сmах метаболитов на 30 % и увеличивает время ее достижения на 3 ч, не изменяя площадь под кривой «концентрация-время» (AUC).
Распределение
Быстро распределяется в тканях. Связь с белками составляет 97 % (сибутрамин) и 94 % (М1 и М2). Равновесная концентрация активных метаболитов в крови достигается в течение 4 суток после начала лечения и примерно в 2 раза превышает концентрацию в плазме крови после приема разовой дозы.
Метаболизм и выведение
Активные метаболиты подвергаются гидроксилированию и конъюгации с образованием неактивных метаболитов, которые экскретируются преимущественно почками. Период полувыведения сибутрамина - 1,1 ч, М1 - 14 ч, М2 - 16 ч.
Фармакокинетика в особых клинических случаях
Имеющиеся в настоящее время данные не указывают на существование клинически значимых различий в фармакокинетике у мужчин и женщин.
Фармакокинетика у пожилых
Фармакокинетика у пожилых здоровых людей (средний возраст 70 лет) аналогична таковой у молодых.
Почечная недостаточность
Почечная недостаточность не оказывает влияния на AUC активных метаболитов M1 и М2, кроме метаболита М2 у пациентов с терминальной стадией почечной недостаточности, находящихся на диализе.
Печеночная недостаточность
У пациентов с печеночной недостаточностью средней степени тяжести после однократного приема сибутрамина AUC активных метаболитов М1 и М2 на 24 % выше, чем у здоровых людей.
с индексом массы тела (ИМТ) более 30 кг/м2 (алиментарное ожирение);
с ИМТ 27 кг/м2 и более в сочетании с сахарным диабетом 2 типа и дислипидемией;
повышенная чувствительность к компонентам препарата;
диабетический кетоацидоз, диабетическая прекома, диабетическая кома;
нарушение функции почек (клиренс креатинина (КК) менее 45 мл/мин);
нарушение функции печени;
острые состояния, при которых имеется риск развития нарушения функции почек: дегидратация (при диарее, рвоте), тяжелые инфекционные заболевания, шок;
сердечно-сосудистые заболевания (в анамнезе и в настоящее время): ишемическая болезнь сердца (инфаркт миокарда (ИМ), стенокардия), хроническая сердечная недостаточность в стадии декомпенсации, окклюзирующие заболевания периферических артерий, тахикардия, аритмия, цереброваскулярные заболевания (инсульт, транзиторные нарушения мозгового кровообращения);
неконтролируемая артериальная гипертензия (артериальное давление (АД) выше 145/90 мм рт. ст.) (см. также раздел «Особые указания»);
клинически выраженные проявления острых и хронических заболеваний, которые могут привести к развитию тканевой гипоксии (в т.ч. дыхательная недостаточность, сердечная недостаточность, острый ИМ);
хронический алкоголизм, острое отравление этанолом;
тиреотоксикоз;
доброкачественная гиперплазия предстательной железы;
феохромоцитома;
закрытоугольная глаукома;
обширные хирургические операции и травмы (когда показано проведение инсулинотерапии);
лактоацидоз (в т.ч. в анамнезе);
установленная фармакологическая или наркотическая зависимость;
беременность и период грудного вскармливания;
возраст до 18 лет и старше 65 лет;
период менее 48 ч до и в течение 48 ч после проведения радиоизотопных или рентгенологических исследований с введением йодо- содержащего контрастного средства;
соблюдение гипокалорийной диеты (менее 1000 ккал/сут);
наличие органических причин ожирения (например, гипотиреоз);
серьезные нарушения питания – нервная анорексия или нервная булимия;
психические заболевания;
синдром Жиль де ля Туретта (генерализованные тики);
одновременный прием ингибиторов МАО (например, фентермина, фенфлурамина, дексфенфлурамина, этиламфетамина, эфедрина) или их применение в течение 2-х недель до приема сибутрамина и 2-х недель после окончания его приема; других препаратов, действующих на центральную нервную систему, ингибирующих обратный захват серотонина (например, антидепрессантов, нейролептиков); снотворных препаратов, содержащих триптофан, а также других препаратов центрального действия для снижения массы тела или лечения психических расстройств.
Поскольку до настоящего времени не имеется достаточно убедительного количества исследований в отношении безопасности воздействия сибутрамина на плод, данный препарат противопоказан в период беременности. Женщины, находящиеся в репродуктивном возрасте, во время приема препарата Редуксин® Форте должны пользоваться контрацептивными средствами.
Противопоказано применение препарата Редуксин® Форте во время грудного вскармливания.Препарат Редуксин® Форте назначается внутрь 1 раз в сутки. Дозу устанавливают индивидуально в зависимости от переносимости и клинической эффективности.
Рекомендуется начальная доза 850 мг+10 мг в сутки.
Препарат следует принимать утром, не разжевывая и запивая достаточным количеством жидкости (стакан воды). Препарат можно принимать как натощак, так и сочетать с приемом пищи. Увеличение дозы до 850 мг+15 мг возможно, если в течение месяца не достигнуто снижение массы тела на 2 кг и более, но не ранее, чем через 4 недели от начала лечения.
Лечение препаратом Редуксин® Форте не должно продолжаться более 3 месяцев у пациентов, которым в течение 3-х месяцев лечения не удается достигнуть 5 % уровня снижения массы тела от исходного уровня при приеме препарата в максимальной дозе.
У пациентов с ожирением без дополнительных нарушений углеводного обмена рекомендован прием препарата Редуксин® Форте в течение 6 месяцев для выработки правильных привычек питания и удержания достигнутого результата по снижению массы тела. Лечение препаратом Редуксин® Форте не следует продолжать, если при дальнейшей терапии после достигнутого снижения массы тела, пациент вновь прибавляет в массе тела 3 кг и более.
Рекомендуется длительный прием препарата для снижения массы тела на 5–10% и удержания результата, что позволяет уменьшить риски для здоровья, а также улучшить течение заболеваний, ассоциированных с ожирением.
Лечение препаратом Редуксин® Форте должно осуществляться в комплексе с диетой и физическими упражнениями под контролем врача, имеющего практический опыт лечения ожирения.
Длительность непрерывного лечения не должна превышать 1 года.
Определение частоты побочных эффектов: очень часто (³1/10), часто (³1/100, <1/10), нечасто (³1/1000, <1/100), редко (³1/10 000,
<1/1000), очень редко (<1/10 000). Побочное действие представлено в порядке снижения значимости.
Метформин
Нарушения со стороны обмена веществ и питания: очень редко – лактоацидоз; при длительном применении возможно снижение всасывания витамина B12. Снижение концентрации витамина В12 необходимо принимать во внимание у пациентов с мегалобластной анемией.
Нарушения со стороны нервной системы: часто – нарушение вкуса.
Нарушения со стороны желудочно-кишечного тракта: очень часто – тошнота, рвота, диарея, боль в животе, отсутствие аппетита. Наиболее часто эти симптомы возникают в начальный период лечения и в большинстве случаев спонтанно проходят. Медленное увеличение дозы может улучшить желудочно-кишечную переносимость.
Нарушения со стороны печени и желчевыводящих путей: очень редко – нарушение функции печени, гепатит, после отмены метформина эти нежелательные явления полностью исчезают.
Нарушения со стороны кожи и подкожной клетчатки: очень редко – кожные реакции, такие как эритема, зуд, сыпь.
Сибутрамин
Чаще всего побочные эффекты возникают в начале лечения (в первые 4 недели). Их выраженность и частота с течением времени ослабевают. Побочные эффекты имеют, в целом, нетяжелый и обратимый характер.
Нарушения со стороны нервной системы: очень часто – сухость во рту и бессонница, часто – головная боль, головокружение, беспокойство, парестезия, а так же изменение вкуса.
Нарушения со стороны сердца: часто – тахикардия, ощущение сердцебиения.
Нарушения со стороны сосудов: повышение артериального давления, вазодилатация. Наблюдается умеренный подъем артериального давления в покое на 1-3 мм рт. ст. и умеренное увеличение пульса на 3-7 ударов в минуту. В отдельных случаях не исключаются более выраженное повышение артериального давления и увеличение частоты сердечных сокращений. Клинически значимые изменения артериального давления и пульса регистрируются преимущественно в начале лечения (в первые 4-8 недель). Нарушения со стороны желудочно-кишечного тракта: очень часто – потеря аппетита и запор, часто – тошнота и обострение геморроя. При склонности к запорам в первые дни необходим контроль за эвакуаторной функцией кишечника. При возникновении запора прием прекращают и принимают слабительное.
Нарушения со стороны кожи и подкожных тканей: часто – повышенное потоотделение.
В единичных случаях при лечении сибутрамином описаны следующие нежелательные клинически значимые явления: дисменорея, отеки, гриппоподобный синдром, зуд кожи, боль в спине, боль в животе, парадоксальное повышение аппетита, жажда, ринит, депрессия, сонливость, эмоциональная лабильность, тревожность, раздражительность, нервозность, острый интерстициальный нефрит, кровотечения, пурпура Шенлейн-Геноха (кровоизлияния в кожу), судороги, тромбоцитопения, транзиторное повышение активности «печеночных» ферментов в крови.
Применение препарата Редуксин® Форте у пациентов с повышенным артериальным давлением: см. разделы «Противопоказания» и «Особые указания».
В ходе постмаркетинговых исследований сибутрамина были описаны дополнительные побочные реакции, перечисленные ниже по системам органов:
Нарушения со стороны иммунной системы: реакции гиперчувствительности (от умеренных высыпаний на коже и крапивницы до ангионевротического отека (отека Квинке) и анафилаксии).
Нарушения психики: психоз, состояния суицидально направленного мышления, суицид и мания. При возникновении подобных состояний препарат необходимо отменить.
Нарушения со стороны нервной системы: судороги, кратковременные нарушения памяти. Нарушения со стороны органа зрения: затуманивание зрения («пелена перед глазами»). Нарушения со стороны сердца: мерцательная аритмия.
Нарушения со стороны желудочно-кишечного тракта: диарея, рвота.
Нарушения со стороны кожи и подкожной клетчатки: алопеция.
Нарушения со стороны почек и мочевыводящих путей: задержка мочи.
Нарушения со стороны половых органов и молочной железы: нарушения эякуляции/оргазма, импотенция, нарушение менструального цик ла, маточные кровотечения.
Метформин
Симптомы: при применении метформина в дозе 85 г (в 42,5 раз превышающей максимальную суточную дозу) не наблюдалось гипогликемии, однако отмечалось развитие лактоацидоза.
Значительная передозировка или сопряженные факторы риска могут привести к развитию лактоацидоза.
Лечение: в случае появления признаков лактоацидоза лечение препаратом необходимо немедленно прекратить, пациента срочно госпитализировать и, определив концентрацию лактата, уточнить диагноз. Наиболее эффективным мероприятием по выведению из организма лактата и метформина является гемодиализ. Проводят так же симптоматическое лечение.
Сибутрамин
Имеются крайне ограниченные данные по поводу передозировки сибутрамина. Наиболее часто встречающиеся неблагоприятные реакции, связанные с передозировкой: тахикардия, повышение артериального давления, головная боль, головокружение. Следует известить своего лечащего врача в случае предполагаемой передозировки.
Лечение: какого-либо специального лечения и специфических антидотов не существует. Необходимо выполнять общие мероприятия: обеспечить свободное дыхание, наблюдать за состоянием сердечно-сосудистой системы, а так же при необходимости осуществить поддерживающую симптоматическую терапию. Своевременное применение активированного угля, а так же промывание желудка может уменьшить поступление сибутрамина в организм. Пациентам с повышенным артериальным давлением и тахикардией можно назначить β-адреноблокаторы. Эффективность форсированного диуреза или гемодиализа не установлена. При передозировке следует немедленно отменить прием препарата Редуксин® Форте.
Метформин
Противопоказанные комбинации
Йодсодержащие рентгеноконтрастные средства: на фоне функциональной почечной недостаточности у больных сахарным диабетом радиологическое исследование с применением йодсодержащих рентгеноконтрастных средств может вызывать развитие лактоацидоза. Лечение метформином необходимо отменить в зависимости от функции почек за 48 ч до или на время рентгенологического исследования с применением йодсодержащих рентгеноконтрастных средств и не возобновлять ранее 48 ч после исследования, при условии, что в ходе обследования почечная функция была признана нормальной.
Нерекомендуемые комбинации
Алкоголь: при острой алкогольной интоксикации увеличивается риск развития лактоацидоза, особенно в случаях:
недостаточного питания, соблюдения низкокалорийной диеты;
печёночной недостаточности.
Во время приема препарата следует избегать приема алкоголя и лекарственных средств, содержащих этанол.
Комбинации, требующие осторожности
Даназол: не рекомендуется одновременный прием даназола во избежание гипергликемического действия последнего. При необходимости лечения даназолом и после прекращения приема последнего требуется коррекция дозы метформина под контролем концентрации глюкозы в крови.
Хлорпромазин: при приеме в больших дозах (100 мг в день) повышает концентрацию глюкозы в крови, снижая высвобождение инсулина. При лечении нейролептиками и после прекращения приема последних требуется коррекция дозы препарата под контролем концентрации глюкозы в крови.
Глюкокортикостероиды (ГКС) системного и местного действия снижают толерантность к глюкозе, повышают концентрацию глюкозы в крови, иногда вызывая кетоз. При лечении ГКС и после прекращения приема последних требуется коррекция дозы метформина под контролем концентрации глюкозы в крови.
Диуретики: одновременный прием «петлевых» диуретиков может привести к развитию лактоацидоза из-за возможной функциональной почечной недостаточности. Не следует назначать метформин, если КК ниже 60 мл/мин.
Назначаемые в виде инъекций бета2-адреномиметики: повышают концентрацию глюкозы в крови вследствие стимуляции бета2-адренорецепторов. В этом случае необходим контроль концентрации глюкозы в крови. При необходимости рекомендуется назначение инсулина.
При одновременном применении вышеперечисленных лекарственных средств может потребоваться более частый контроль кон - центрации глюкозы в крови особенно в начале лечения. При необходимости доза метформина может быть скорректирована в процессе лечения и после его прекращения.
Ингибиторы ангиотензинпревращающего фермента и другие гипотензивные лекарственные средства могут снижать концентрацию глюкозы в крови. При необходимости следует скорректировать дозу метформина.
Гипогликемическое действие метформина могут снижать фенотиазиды, глюкагон, эстрогены, пероральные контрацептивы, фенитоин, симпатомиметики, никотиновая кислота, изониазид, блокаторы «медленных» кальциевых каналов, левотироксин натрия. Одновременное применение с циметидином снижает скорость выведения метформина, что может приводить к развитию лактоацидоза. У здоровых добровольцев при одновременном применении метформина и пропранолола, а так же при применении метформина и ибупрофена не наблюдалось изменения их фармакокинетических показателей. Метформин может снижать действие антикоагулянтов непрямого действия.
Субстраты транспортера органических катионов 1 и 2 (ОСТ1 И ОСТ2)
Метформин является субстратом органических катионов ОСТ1 и ОСТ2. При совместном применении с метформином:
ингибиторы ОСТ1 (такие как верапамил) могут снизить гипогликемическое действие метформина.
индукторы ОСТ1 (такие как рифампицин) могут увеличить всасывание метформина в желудочно-кишечном тракте и усилить его гипогликемическое действие.
ингибиторы ОСТ2 (такие как циметидин, долутегравир, ранолазин, триметоприм, вандетаниб, изавуконазол) могут снизить выведение метформина почками и привести к увеличению его концентрации в плазме крови.
Ингибиторы ОСТ1 и ОСТ2 (такие как кризотиниб, олапарид) могут снизить гипогликемическое действие метформина.
При одновременном применении метформина с производными сульфонилмочевины, инсулином, акарбозой, салицилатами возможно развитие гипогликемии.
Нифедипин повышает абсорбцию и Cmax метформина.
Катионные лекарственные средства (амилорид, дигоксин, морфин, прокаинамид, хинидин, хинин, ранитидин, триамтерен, триметоприм и ванкомицин), секретирующиеся в почечных канальцах, конкурируют с метформином за канальцевые транспортные системы и могут приводить к увеличению его Сmax.
Сибутрамин
Ингибиторы микросомального окисления, в т.ч. ингибиторы изофермента CYP3A4 (кетоконазол, эритромицин, циклоспорин и др.) повышают в плазме крови концентрации метаболитов сибутрамина с увеличением частоты сердечных сокращений и клинически несущественным увеличением интервала QT.
Рифампицин, антибиотики из группы макролидов, фенитоин, карбамазепин, фенобарбитал и дексаметазон могут ускорять метаболизм сибутрамина.
Одновременное применение нескольких препаратов, повышающих содержание серотонина в плазме крови, может привести к развитию серьезного взаимодействия. Так называемый серотониновый синдром может развиться в редких случаях при одновременном применении сибутрамина с селективными ингибиторами обратного захвата серотонина (препаратами для лечения депрессии), с некоторыми препаратами для лечения мигрени (суматриптан, дигидроэрготамин), с сильнодействующими анальгетиками (пентазоцин, петидин, фентанил), или противокашлевыми препаратами (декстрометорфан). Сибутрамин не влияет на действие пероральных контрацептивных средств.
При одновременном приеме сибутрамина и алкоголя не было отмечено усиления негативного действия алкоголя. Однако алкоголь абсолютно не сочетается с рекомендуемыми при приеме сибутрамина диетическими мероприятиями.
При одновременном применении с сибутрамином других препаратов, влияющих на гемостаз или функцию тромбоцитов, увеличивается риск развития кровотечений. Лекарственное взаимодействие при одновременном применении сибутрамина с препаратами, повышающими артериальное давление и частоту сердечных сокращений, в настоящее время недостаточно полно изучено. Эта группа препаратов включает деконгестанты, противокашлевые, противопростудные и противоаллергические препараты, в состав которых входят эфедрин или псевдоэфедрин. Поэтому в случаях одновременного приема этих препаратов с сибутрамином следует соблюдать осторожность.
Совместное применение сибутрамина с препаратами для снижения массы тела, действующими на центральную нервную систему, или препаратами для лечения психических расстройств противопоказано.
Лактоацидоз
Лактоацидоз является редким, но серьезным (высокая смертность при отсутствии неотложного лечения) осложнением, которое может возникнуть из-за кумуляции метформина. Случаи лактоацидоза при приеме метформина возникали в основном у пациентов сахарным диабетом с выраженной почечной недостаточностью.
Следует учитывать и другие сопряженные факторы риска, такие как декомпенсированный сахарный диабет, кетоз, продолжительное голодание, алкоголизм, печеночная недостаточность и любое состояние, связанное с выраженной гипоксией. Это может помочь снизить частоту случаев возникновения лактоацидоза.
Следует учитывать риск развития лактоацидоза при появлении неспецифических признаков, таких как мышечные судороги, сопровождающиеся диспепсическими симптомами, болью в животе и выраженной астенией. Лактоацидоз характеризуется ацидотической одышкой, болью в животе и гипотермией с последующей комой. Диагностическими лабораторными показателями являются снижение рН крови (менее 7,25), содержание лактата в плазме крови свыше 5 ммоль/л, повышенные анионный промежуток и отношение лактат/пируват. При подозрении на метаболический ацидоз необходимо прекратить прием препарата и немедленно обратиться к врачу.
Хирургические операции
Применение препарата Редуксин® Форте должно быть прекращено за 48 ч до проведения плановых хирургических операций и может быть продолжено не ранее, чем через 48 ч после при условии, что в ходе обследования почечная функция была признана нормальной.
Функция почек
Поскольку метформин выводится почками, перед началом приема препарата Редуксин® Форте и регулярно в последующем, необходимо определять КК: не реже 1 раза в год у пациентов с нормальной функцией почек, и 2-4 раза в год у пациентов пожилого возраста, а также у пациентов с КК на нижней границе нормы.
Следует проявлять особую осторожность при возможном нарушении функций почек у пациентов пожилого возраста, при одновременном применении гипотензивных препаратов, диуретиков или нестероидных противовоспалительных препаратов.
Пациентам рекомендуется продолжать соблюдать диету с равномерным употреблением углеводов в течение дня. Пациентам с избыточной массой тела рекомендуется продолжать соблюдать гипокалорийную диету (но не менее 1000 ккал/сут).
Рекомендуется регулярно проводить стандартные лабораторные анализы для контроля сахарного диабета.
Рекомендуется проявлять осторожность при применении препарата Редуксин® Форте в комбинации с инсулином или другими гипогликемическими средствами (в т.ч. производными сульфонилмочевины, репаглинидом).
Лечение препаратом Редуксин® Форте должно осуществляться в рамках комплексной терапии по снижению массы тела под контролем врача, имеющего практический опыт лечения ожирения. Комплексная терапия включает в себя как изменение диеты и образа жизни, так и увеличение физической активности. Важным компонентом терапии является создание предпосылок к стойкому изменению пищевого поведения и образа жизни, которые необходимы для сохранения достигнутого снижения массы тела и после отмены медикаментозной терапии. Пациентам необходимо в рамках терапии препаратом Редуксин® Форте изменить свой жизненный уклад и привычки таким образом, чтобы после завершения лечения обеспечить сохранение достигнутого уменьшения массы тела. Пациенты должны четко представлять себе, что несоблюдение этих требований приведет к повторному увеличению массы тела и повторным обращениям к лечащему врачу.
У пациентов, принимающих препарат Редуксин® Форте, необходимо измерять артериальное давление и частоту сердечных сокращений. В первые 3 месяца лечения эти параметры следует контролировать каждые 2 недели, а затем ежемесячно. Если во время двух визитов подряд выявляется увеличение частоты сердечных сокращений в покое ≥10 ударов в минуту или систолического/диастолического давления ≥10 мм рт. ст., необходимо прекратить лечение. У пациентов с артериальной гипертензией, у которых на фоне гипотензивной терапии артериальное давление выше 145/90 мм рт. ст., этот контроль должен проводиться особенно тщательно и, при необходимости, через более короткие интервалы. У пациентов, у которых артериальное давление дважды при повторном измерении превышало 145/90 мм рт. ст., лечение препаратом Редуксин® Форте должно быть приостановлено (см. раздел «Побочное действие», подразделы «Нарушения со стороны сердца», «Нарушения со стороны сосудов»).
У пациентов с синдромом апноэ во сне необходимо особенно тщательно контролировать артериальное давление.
Особого внимания требует одновременное назначение препаратов, увеличивающих интервал QT. К этим препаратам относятся блокаторы H1-гистаминовых рецепторов (астемизол, терфенадин); антиаритмические препараты, увеличивающие интервал QT (амиодарон, хинидин, флекаинид, мексилетин, пропафенон, соталол); стимулятор моторики желудочно-кишечного тракта цизаприд; пимозид, сертиндол и трициклические антидепрессанты. Это касается и состояний, которые способны приводить к увеличению интервала QT, таких как гипокалиемия и гипомагниемия (см. раздел «Взаимодействие с другими лекарственными средствами»).
Интервал между приемом ингибиторов МАО (в т.ч. фуразолидона, прокарбазина, селегилина) и препаратом Редуксин® Форте должен составлять не менее 2 недель.
Хотя не установлена связь между приемом сибутрамина и развитием первичной легочной гипертензии, однако, учитывая общеизвестный риск препаратов данной группы, при регулярном медицинском контроле необходимо особое внимание обращать на такие симптомы, как прогрессирующее диспноэ (нарушение дыхания), боль в грудной клетке и отеки на ногах.
При пропуске дозы препарата Редуксин® Форте не следует принимать в следующий прием двойную дозу препарата, рекомендовано продолжать дальнейший прием препарата по предписанной схеме.
При совместном приеме сибутрамина и других ингибиторов обратного захвата серотонина существует повышенный риск развития кровотечений. У пациентов, предрасположенных к кровотечениям, а также принимающих препараты, влияющие на гемостаз или функцию тромбоцитов, сибутрамин следует применять с осторожностью.
Хотя клинические данные о привыкании к сибутрамину отсутствуют, следует выяснить, не было ли в анамнезе пациента случаев лекарственной зависимости, и обратить внимание на возможные признаки злоупотребления лекарственными препаратами.
Таблетки, покрытые пленочной оболочкой, 850 мг + 10 мг; 850 мг + 15 мг.
По 5, 7, 10 или 15 таблеток, покрытых пленочной оболочкой, в контурную ячейковую упаковку из пленки поливинилхлоридной и фольги алюминиевой печатной лакированной.
По 10, 15, 30, 60, 75, 100, 120 или 180 таблеток, покрытых пленочной оболочкой, в банку из полиэтилентерефталата для лекарственных средств, укупоренную крышкой навинчиваемой с контролем первого вскрытия из полиэтилена низкого давления. Свободное пространство в банке заполняют ватой медицинской гигроскопической.
Одну банку или 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10 контурных ячейковых упаковок вместе с инструкцией по применению помещают в картонную упаковку (пачку).
В защищенном от света месте при температуре не выше 25 °С. Препарат следует хранить в местах, недоступных для детей.
Сибутрамин относится к Списку сильнодействующих веществ, утвержденному Постановлением Правительства РФ от 29.12.2007 г. № 964.
3 года.
Не применять по истечении срока годности.
Отпускают по рецепту.
АО «Биохимик», Россия.
Юридический адрес: Россия, 430030, Республика Мордовия, г. Саранск, ул. Васенко, 15А.
Адрес места производства: Россия, 430030, Республика Мордовия, г. Саранск, ул. Васенко, 15А. Телефон: (8342) 38-03-68.
E-mail: biohimic@biohimic.ru, www.biohimik.ru
ООО «ПРОМОМЕД РУС», Россия.
Адрес: Россия, 129090, г. Москва, пр-т Мира, д. 13, стр. 1. Тел.: 8-800-777-86-04 (бесплатно), 8 (495) 640-25-28.
E-mail: reception@promo-med.ru
№ ЛС-002110 от 29.02.2012
Сибутрамин + целлюлоза микрокристаллическая
Форма выпуска «Редуксина®» – капсулы10мг (сибутрамина гидрохлорида моногидрат 10 мг + целлюлоза микрокристаллическая – 158,5 мг), капсулы 15 мг (сибутрамина гидрохлорида моногидрат 15 мг + целлюлоза микрокристаллическая – 153,5 мг).
«Редуксин®» – комбинированный препарат, предназначенныйдля правильного снижения веса и обусловленный компонентами, входящим в состав: сибутрамин и МКЦ. Он является одним из вспомогательных способов в комплексном подходе к снижению веса.
Снижение массы тела при алиментарном ожирении с индексом массы тела (ИМТ) 30 кг/м2 и более или при алиментарном ожирении с индексом массы тела 27 кг/м2 и более при наличии сахарного диабета 2 типа и дислипидемии.
Следует назначать препарат при следующих состояниях: аритмии в анамнезе, хронической недостаточности кровообращения, заболеваниях коронарных артерий (в т.ч. в анамнезе), кроме ишемической болезни сердца (ИМ, стенокардии); глаукоме, кроме закрытоугольной глаукомы, холелитиазе, артериальной гипертензии (контролируемой и в анамнезе), неврологических нарушениях, включая задержку умственного развития и судороги (в т.ч. в анамнезе), эпилепсии, нарушении функции печени и/или почек легкой и средней степени тяжести, моторных и вербальных тиках в анамнезе, склонности к кровотечению, нарушению свертываемости крови, приеме препаратов, влияющих на гемостаз или функцию тромбоцитов.
Редуксин принимается внутрь 1 раз в сутки. Дозу устанавливают индивидуально, в зависимости от переносимости и клинической эффективности.
Рекомендуется начальная доза 10 мг/сутки. Капсулы следует принимать утром, не разжевывая и запивая достаточным количеством жидкости (стакан воды). Препарат можно принимать как натощак, так и сочетать с приемом пищи. Если в течение 4-х недель от начала лечения не достигнуто снижение массы тела более 2 кг, то доза увеличивается до 15 мг/сут. Лечение препаратом Редуксин® не должно продолжаться более 3 месяцев у пациентов, которые недостаточно хорошо реагируют на терапию, т.е. которым в течение 3-х месяцев лечения не удается достичь снижения массы тела на 5 % от исходного показателя. Лечение не следует продолжать, если при дальнейшей терапии, после достигнутого снижения массы тела, пациент вновь прибавляет в массе тела 3 кг и более. Длительность лечения не должна превышать 1 года, поскольку в отношении более продолжительного периода приема сибутрамина данные об эффективности и безопасности отсутствуют.
Лечение препаратом Редуксин® должно осуществляться в комплексе с диетой и физическими упражнениями под контролем врача, имеющего практический опыт лечения ожирения.
Чаще всего побочные эффекты возникают в первые 4 лечения. Их выраженность и частота с течением времени ослабевают и носят, в целом, нетяжелый и обратимый характер. Очень часто или часто возникают сухость во рту, бессонница, головная боль, головокружение, беспокойство, парестезии, а также изменение вкуса, тахикардия, ощущение сердцебиения, повышение артериального давления, вазодилатация, потеря аппетита и запор, часто тошнота и обострение геморроя, повышенное потоотделение.
Симптомы: имеются крайне ограниченные данные по поводу передозировки сибутрамина. Наиболее часто встречающиеся неблагоприятные реакции, связанные с передозировкой, – тахикардия, повышение АД, головная боль, головокружение. Пациенту следует известить своего лечащего врача в случае предполагаемой передозировки и придерживаться его показаний.
Лечение: какого-либо специального лечения и специфических антидотов не существует. Необходимо выполнять общие мероприятия: обеспечить свободное дыхание, наблюдать за состоянием ССС, а также при необходимости осуществлять поддерживающую симптоматическую терапию. Своевременное назначение активированного угля, а также промывание желудка могут уменьшить поступление сибутрамина в организм. Пациентам с повышенном АД и тахикардиейназначаются бета-адреноблокаторы. Эффективность форсированного диуреза или гемодиализа не установлена.Сколько и какой именно препарат принимать,решает только лечащий врач.
ООО «ОЗОН», Россия, 445351, Самарская об л., г. Жигулевск, ул. Песочная, 11
Тел./факс: (84862) 3-41-09.
В 1 мл препарата содержится:
Действующее вещество: семаглутид 1,34 мг;
Вспомогательные вещества: натрия гидрофосфата дигидрат, фенол, пропиленгликоль, 1 М раствор натрия гидроксида или 1 М раствор хлористоводородной кислоты, вода для инъекций.
Для шприц-ручки Квинсента® 0,25/0,5 мг/доза:
В одной предварительно заполненной шприц-ручке содержится 2 мг семаглутида/1,5 мл раствора лекарственного препарата.
Для шприц-ручки Квинсента® 0,25/0,5/1 мг/доза:
В одной предварительно заполненной шприц-ручке содержится 4 мг семаглутида/3 мл раствора лекарственного препарата.
Препарат Квинсента® содержит в качестве действующего вещества семаглутид, который представляет собой химически синтезированный пептид и является агонистом рецепторов ГПП-1 (ГПП-1Р). Семаглутид – это аналог человеческого ГПП-1, имеющий 94 % гомологий с аминокислотной последовательностью эндогенного ГПП-1.
Семаглутид действует как агонист ГПП-1Р, который селективно связывается и активирует ГПП-1Р. ГПП-1Р служит мишенью для нативного ГПП-1.
ГПП-1 является физиологическим гормоном, оказывающим одновременно несколько эффектов, включая регуляцию концентрации глюкозы и аппетита, а также влияние на сердечно-сосудистую систему (ССС). Влияние на концентрацию глюкозы и аппетит специфически опосредовано ГПП-1Р, расположенными в поджелудочной железе и головном мозге. Фармакологические концентрации семаглутида снижают концентрацию глюкозы крови и массу тела посредством сочетания эффектов, описанных ниже. ГПП-1Р представлены также в специфических областях сердца, сосудов, иммунной системы и почек, где их активация может оказывать сердечно-сосудистые (СС) и микроциркуляторные эффекты.
В отличие от нативного ГПП-1, длительный период полувыведения семаглутида (около 1 недели) позволяет применять его подкожно (п/к) 1 раз в неделю. Связывание с альбумином является основным механизмом длительного действия семаглутида, что приводит к снижению выведения его почками и защищает от метаболического распада. Кроме того, семаглутид стабилен в отношении расщепления ферментом дипептидилпептидазой-4 (ДПП-4).
Семаглутид снижает концентрацию глюкозы крови посредством глюкозозависимой стимуляции секреции инсулина и подавления секреции глюкагона. Таким образом, при повышении концентрации глюкозы крови происходит стимуляция секреции инсулина и подавление секреции глюкагона. Механизм снижения уровня гликемии включает также небольшую задержку опорожнения желудка в ранней постпрандиальной фазе. Во время гипогликемии семаглутид уменьшает секрецию инсулина и не снижает секрецию глюкагона.
Семаглутид снижает общую массу тела и массу жировой ткани, уменьшая потребление энергии. Данный механизм затрагивает общее снижение аппетита, включая усиление сигналов насыщения и ослабление сигналов голода, а также улучшение контроля потребления пищи и снижение тяги к пище. Также снижается инсулинорезистентность. Помимо этого, семаглутид снижает предпочтение к выбору пищи с высоким содержанием жиров. В исследованиях на животных было показано, что семаглутид поглощается специфическими областями головного мозга и усиливает ключевые сигналы насыщения, и ослабляет ключевые сигналы голода. Воздействуя на изолированные участки тканей головного мозга, семаглутид активирует нейроны, связанные с чувством сытости, и подавляет нейроны, связанные с чувством голода.
В клинических исследованиях (КИ) семаглутид оказывал положительное влияние на липиды плазмы крови, снижал систолическое артериальное давление (АД) и уменьшал воспаление.
В исследованиях на животных семаглутид подавлял развитие атеросклероза, предупреждая дальнейшее развитие аортальных бляшек и уменьшая воспаление в бляшках.
Фармакодинамика
Оценка фармакодинамических параметров проводилась после 12 недель терапии (включая период увеличения дозы) в равновесной концентрации семаглутида 1 мг 1 раз в неделю.
Уровень гликемии натощак и постпрандиальный уровень гликемии
Семаглутид снижает концентрацию глюкозы натощак и концентрацию постпрандиальной глюкозы (ППГ). По сравнению с плацебо терапия семаглутидом в дозе 1 мг у пациентов с сахарным диабетом 2 типа (СД2) приводила к снижению концентрации глюкозы с точки зрения абсолютного изменения от исходного значения (ммоль/л) и относительного снижения по сравнению с плацебо (%) в отношении: концентрации глюкозы натощак (1,6 ммоль/л; 22 %); концентрации глюкозы через 2 часа после приема пищи (4,1 ммоль/л; 37 %); средней суточной концентрации глюкозы (1,7 ммоль/л; 22 %) и постпрандиальных пиков концентрации глюкозы за 3 приема пищи (0,6–1,1 ммоль/л). Семаглутид снижал концентрацию глюкозы натощак после введения первой дозы.
Функция бета-клеток поджелудочной железы и секреция инсулина
Семаглутид улучшает функцию бета-клеток поджелудочной железы. После внутривенного струйного введения глюкозы пациентам с СД2 семаглутид по сравнению с плацебо улучшал первую и вторую фазу инсулинового ответа с трехкратным и двукратным повышением, соответственно, и увеличивал максимальную секреторную активность бета-клеток поджелудочной железы после теста стимуляции аргинином. Кроме того, по сравнению с плацебо терапия семаглутидом увеличивает концентрацию инсулина натощак.
Секреция глюкагона
Семаглутид снижает концентрацию глюкагона натощак и постпрандиальную концентрацию глюкагона. У пациентов с СД2 семаглутид приводит к относительному снижению концентрации глюкагона по сравнению с плацебо: концентрации глюкагона натощак (8–21 %), постпрандиального глюкагонового ответа (14–15 %) и средней суточной концентрации глюкагона (12 %).
Глюкозозависимая секреция инсулина и глюкозозависимая секреция глюкагона
Семаглутид снижал высокую концентрацию глюкозы в крови, стимулируя секрецию инсулина и снижая секрецию глюкагона глюкозозависимым способом. Скорость секреции инсулина после введения семаглутида пациентам с СД2 была сопоставима с таковой у здоровых добровольцев.
Во время индуцированной гипогликемии семаглутид по сравнению с плацебо не изменял контррегуляторный ответ повышения концентрации глюкагона, а также не усугублял снижение концентрации С-пептида у пациентов с СД2.
Опорожнение желудка
Семаглутид вызывал небольшую задержку раннего постпрандиального опорожнения желудка, тем самым снижая скорость поступления ППГ в кровь.
Масса тела и состав тела
Наблюдалось большее снижение массы тела при применении семаглутида по сравнению с изученными препаратами сравнения (плацебо, ситаглиптином, эксенатидом замедленного высвобождения (ЗВ), дулаглутидом и инсулином гларгин) (см. раздел «Клиническая эффективность и безопасность»). Потеря массы тела при применении семаглутида происходила преимущественно за счет потери жировой ткани, превышающей потерю мышечной массы в 3 раза.
Аппетит, потребление калорий и выбор продуктов питания
По сравнению с плацебо семаглутид снизил потребление калорий на 18–35 % во время трех последовательных приемов пищи ad libitum за счет подавления аппетита (как натощак, так и после приема пищи), улучшая контроль потребления пищи, ослабляя тягу к еде, особенно с высоким содержанием жиров.
Липиды натощак и постпрандиальные липиды
По сравнению с плацебо семаглутид снижал концентрации триглицеридов и холестерина липопротеинов очень низкой плотности (ЛПОНП) натощак на 12 % и 21 %, соответственно. Постпрандиальное увеличение концентрации триглицеридов и холестерина ЛПОНП в ответ на прием пищи с высоким содержанием жиров снизилось более чем на 40 %.
Электрофизиология сердца (ЭФс)
Действие семаглутида на процесс реполяризации в сердце было протестировано в исследовании ЭФс. Применение семаглутида в дозах, превышающих терапевтические (в равновесной концентрации до 1,5 мг), не приводило к удлинению скорректированного интервала QT.
Клиническая эффективность и безопасность
Как улучшение гликемического контроля, так и снижение СС заболеваний и смертности являются неотъемлемой частью лечения СД2.
Эффективность и безопасность семаглутида в дозах 0,5 мг и 1 мг оценивались в шести рандомизированных контролируемых КИ 3а фазы. Из них в пяти КИ в качестве основной цели оценивали эффективность гликемического контроля, в то время как в одном КИ оценивали в качестве основной цели СС исходы. В дополнение были проведены два КИ семаглутида 3 фазы с участием японских пациентов.
В дополнение было проведено исследование 3b фазы для сравнения эффективности и безопасности семаглутида в дозах 0,5 мг и 1 мг один раз в неделю с дулаглутидом 0,75 мг и 1,5 мг один раз в неделю, соответственно. Также было проведено КИ 3b фазы с целью изучения эффективности и безопасности семаглутида в качестве дополнения к лечению ингибитором натрийзависимого переносчика глюкозы 2 типа (SGLT2).
Терапия семаглутидом продемонстрировала устойчивые, статистически превосходящие и клинически значимые улучшения показателя HbA1с и снижение массы тела на срок до 2 лет по сравнению с плацебо и лечением с активным контролем (ситаглиптином, инсулином гларгин, эксенатидом ЗВ и дулаглутидом).
Возраст, пол, раса, этническая принадлежность, исходные значения индекса массы тела (ИМТ) и массы тела (кг), длительность сахарного диабета (СД) и почечная недостаточность не повлияли на эффективность семаглутида.
Монотерапия
Монотерапия семаглутидом в дозах 0,5 мг и 1 мг 1 раз в неделю в течение
30 недель по сравнению с плацебо приводила к статистически более значимому снижению показателей НbА1с (-1,5 %, -1,6 % против 0 %, соответственно), глюкозы плазмы натощак (ГПН) (-2,5 ммоль/л, -2,3 ммоль/л против -0,6 ммоль/л, соответственно) и массы тела (-3,7 кг, -4,5 кг против
-1,0 кг, соответственно).
Семаглутид по сравнению с ситаглиптином, оба в комбинации с 1–2 пероральными гипогликемическими препаратами (ПГГП) (метформином и/или препаратами группы тиазолидиндиона)
Терапия семаглутидом 0,5 мг и 1 мг 1 раз в неделю в течение 56 недель по сравнению с ситаглиптином привела к устойчивому и статистически более значимому снижению показателей НbА1c (-1,3 %, -1,6 % против -0,5 %, соответственно), ГПН (-2,1 ммоль/л, -2,6 ммоль/л против -1,1 ммоль/л, соответственно) и массы тела (-4,3 кг, -6,1 кг против -1,9 кг, соответственно). Терапия семаглутидом 0,5 мг и 1 мг по сравнению с ситаглиптином значительно снижала систолическое АД от исходного значения в 132,6 мм рт.ст. (-5,1 мм рт.ст., -5,6 мм рт.ст. против -2,3 мм рт.ст., соответственно). Изменений диастолического АД не происходило.
Семаглутид по сравнению с дулаглутидом, оба в комбинации с метформином
Терапия семаглутидом 0,5 мг по сравнению с дулаглутидом 0,75 мг, оба 1 раз в неделю на протяжении 40 недель, привела к устойчивому и статистически превосходящему снижению показателей HbA1с (-1,5 % против -1,1 %), ГПН (- 2,2 ммоль/л против -1,9 ммоль/л) и массы тела (-4,6 кг против -2,3 кг), соответственно.
Терапия семаглутидом 1 мг по сравнению с дулаглутидом 1,5 мг, оба 1 раз в неделю на протяжении 40 недель, привела к устойчивому и статистически превосходящему снижению показателей HbA1с (-1,8 % против -1,4 %), ГПН (- 2,8 ммоль/л против -2,2 ммоль/л) и массы тела (-6,5 кг против -3,0 кг), соответственно.
Семаглутид по сравнению с эксенатидом ЗВ, оба в комбинации с метформином или метформином совместно с производным сульфонилмочевины
Терапия семаглутидом 1 мг 1 раз в неделю на протяжении 56 недель по сравнению с эксенатидом ЗВ 2,0 мг привела к устойчивому и статистически более значимому снижению показателей HbA1с (-1,5 % против -0,9 %), ГПН
(-2,8 ммоль/л против -2,0 ммоль/л) и массы тела (-5,6 кг против -1,9 кг), соответственно.
Семаглутид по сравнению с инсулином гларгин, оба в комбинации с 1–2 ПГГП (монотерапия метформином или метформин с производным сульфонилмочевины)
Терапия семаглутидом в дозах 0,5 мг и 1 мг 1 раз в неделю по сравнению с инсулином гларгин в течение 30 недель привела к статистически более значимому снижению показателей HbA1c (-1,2 %, -1,6 % против -0,8 %, соответственно) и массы тела (-3,5 кг, -5,2 кг против +1,2 кг, соответственно).
Снижение показателя ГПН было статистически более значимым для семаглутида 1 мг по сравнению с инсулином гларгин (-2,7 ммоль/л против -2,1 ммоль/л). Не наблюдалось статистически более значимое снижение показателя ГПН для семаглутида 0,5 мг (-2,0 ммоль/л против -2,1 ммоль/л).
Доля пациентов, у которых наблюдались тяжелые или подтвержденные (< 3,1 ммоль/л) эпизоды гипогликемии, была ниже при применении семаглутида 0,5 мг (4,4 %) и семаглутида 1 мг (5,6 %) по сравнению с инсулином гларгин (10,6 %).
Больше пациентов достигли показателя HbA1с < 7 % без тяжелых или подтвержденных эпизодов гипогликемии и без набора веса при применении семаглутида 0,5 мг (47 %) и семаглутида 1 мг (64 %) по сравнению с инсулином гларгин (16 %).
Семаглутид по сравнению с плацебо, оба в комбинации с базальным инсулином
Терапия семаглутидом в дозах 0,5 мг и 1 мг по сравнению с плацебо в течение 30 недель привела к статистически более значимому снижению показателей HbA1с (-1,4 %, -1,8 % против -0,1 %, соответственно), ГПН (-1,6 ммоль/л, -2,4 ммоль/л против -0,5 ммоль/л, соответственно) и массы тела (-3,7 кг, -6,4 кг против -1,4 кг, соответственно). Частота тяжелых или подтвержденных эпизодов гипогликемии существенно не различалась при применении семаглутида и плацебо. Доля пациентов с показателем HbA1с < 8 % на скрининге, сообщивших о тяжелых или подтвержденных (< 3,1 ммоль/л) эпизодах гипогликемии, была выше при применении семаглутида по сравнению с плацебо и сопоставима у пациентов с показателем HbA1с > 8 % на скрининге.
Семаглутид по сравнению с плацебо в качестве дополнения к терапии ингибитором SGLT2 (в качестве монотерапии или в комбинации с производным сульфонилмочевины или метформином).
Терапия семаглутидом в дозе 1 мг 1 раз в неделю в качестве дополнения к терапии ингибитором SGLT2 (в качестве монотерапии или в комбинации с производным сульфонилмочевины или метформином) по сравнению с плацебо 1 раз в неделю в течение 30 недель привела к статистическому значимому снижению показателей HbA1с (-1,5 %, против -0,1%, соответственно), ГПН (2,2 ммоль/л против 0 ммоль/л, соответственно) и массы тела (-4,7 кг против 0,9 кг, соответственно).
Комбинация с монотерапией производным сульфонилмочевины
На 30-й неделе КИ (см. подраздел «Оценка влияния на ССС») была произведена оценка подгруппы из 123 пациентов, находящихся на монотерапии производным сульфонилмочевины. На 30-й неделе показатель HbA1с снизился на 1,6 % и на 1,5 % при применении семаглутида в дозах 0,5 мг и 1 мг, соответственно, и увеличился на 0,1 % при применении плацебо.
Комбинация с предварительно смешанным инсулином ± 1-2 ПГГП
На 30-й неделе КИ (см. подраздел «Оценка влияния на ССС») была произведена оценка подгруппы из 867 пациентов, находящихся на терапии предварительно смешанным инсулином (в комбинации или без 2-х ПГГП). На 30-й неделе показатель HbA1с снизился на 1,3 % и на 1,8 % при применении семаглутида в дозах 0,5 мг и 1 мг, соответственно, и снизился на 0,4 % при применении плацебо.
Соотношение пациентов, достигших целевого снижения показателя HbA1с
До 79 % пациентов достигли цели лечения в отношении снижения показателя HbA1с < 7 %, и доля таких пациентов была значительно больше при применении семаглутида по сравнению с пациентами, получавшими ситаглиптин, эксенатид ЗВ, инсулин гларгин, дулаглутид и плацебо.
Доля пациентов, достигших показателя HbA1с менее 7 % без тяжелых или подтвержденных эпизодов гипогликемии и без набора веса, была значительно больше при применении семаглутида в дозах 0,5 мг и 1 мг (до 66 % и 74 %, соответственно) по сравнению с пациентами, получавшими ситаглиптин (27 %), эксенатид ЗВ (29 %), инсулин гларгин (16 %), дулаглутид 0,75 мг (44 %) и дулаглутид 1,5 мг (58 %).
Масса тела
Монотерапия семаглутидом 1 мг или терапия в комбинации с
1–2 лекарственными препаратами приводила к статистически большему снижению массы тела (потеря составляла до 6,5 кг) по сравнению с терапией плацебо, ситаглиптином, эксенатидом ЗВ, инсулином гларгин или дулаглутидом. Снижение массы тела было устойчивым на срок до 2-х лет.
После одного года терапии большее количество пациентов достигло потери массы тела ≥ 5 % и ≥ 10 % при применении семаглутида 0,5 мг (46 % и 13 %) и 1 мг (до 62 % и 24 %) по сравнению с пациентами, находившимися на терапии активными препаратами сравнения ситаглиптином и эксенатидом ЗВ (до 18 % и до 4 %).
В КИ длительностью 40 недель большее количество пациентов достигло потери массы тела ≥ 5 % и ≥ 10 % при применении семаглутида 0,5 мг (44 % и 14 %) по сравнению с пациентами, получавшими дулаглутид 0,75 мг (23 % и 3 %). Потери массы ≥ 5 % и ≥ 10 % достигло большее количество пациентов, получавших семаглутид 1 мг (до 63 % и 27 %), по сравнению с пациентами, получавшими дулаглутид 1,5 мг (30 % и 8 %). В СС КИ большее количество пациентов достигло потери массы тела ≥ 5 % и ≥ 10 % при применении семаглутида 0,5 мг (36 % и 13 %) и 1 мг (47 % и 20 %) по сравнению с пациентами, получавшими плацебо 0,5 мг (18 % и 6 %) и 1 мг (19 % и 7 %).
ГПН и постпрандиальное увеличение концентрации глюкозы
Во время всех трех ежедневных приемов пищи семаглутид 0,5 мг и 1 мг показал значительное снижение концентрации ГПН до 2,8 ммоль/л и снижение постпрандиального прироста концентрации глюкозы до 1,2 ммоль/л (разница между значениями до и после еды, полученная после трех приемов пищи) (в дополнение см. раздел «Фармакодинамика»).
Функция бета-клеток поджелудочной железы и инсулинорезистентность
В ходе терапии семаглутидом 0,5 мг и 1 мг произошло улучшение функции бета-клеток поджелудочной железы и уменьшение инсулинорезистентности, что подтверждается оценкой гомеостатических моделей функции бета-клеток поджелудочной железы (НОМА-В) и инсулинорезистентности (HOMA-IR) (в дополнение см. раздел «Фармакодинамика»).
Липиды
Во время КИ семаглутида наблюдалось улучшение профиля липидов крови натощак, преимущественно в группе, получавшей дозу 1 мг (в дополнение см. раздел «Фармакодинамика»).
Оценка влияния на ССС
3297 пациентов с СД2 и высоким СС риском были рандомизированы в двойное слепое КИ длительностью 104 недели с применением семаглутида 0,5 мг или 1 мг 1 раз в неделю либо плацебо, соответственно, в дополнение к стандартной терапии СС заболеваний в течение последующих двух лет.
Терапия семаглутидом привела к снижению на 26 % риска серьезных сердечно-сосудистых событий (СССС), включая смерть по причине СС патологии, инфаркт миокарда (ИМ) без смертельного исхода и инсульт без смертельного исхода. В первую очередь это было обусловлено значительным уменьшением частоты инсульта без смертельного исхода (39 %) и незначительным уменьшением частоты ИМ без смертельного исхода (26 %).
Значительно снизился риск реваскуляризации миокарда или периферических артерий, в то время как риск нестабильной стенокардии, требующей госпитализации, и риск госпитализации по причине сердечной недостаточности снизились незначительно.
Микроциркуляторные исходы включали в себя 158 новых или ухудшившихся случаев нефропатии. Относительный риск (ОР) в отношении времени до возникновения нефропатии (новые случаи развития персистирующей макроальбуминурии, персистирующее удвоение сывороточной концентрации креатинина, необходимость в постоянной заместительной почечной терапии и смерть по причине болезни почек) составил 0,64.
В дополнение к стандартной терапии СС заболеваний терапия семаглутидом в дозах 0,5 мг и 1 мг по сравнению с плацебо, соответственно, в течение 104 недель привела к значительному и устойчивому снижению от исходных значений показателя НbА1c (-1,1 % и -1,4 % против -0,4 %
и -0,4 %, соответственно).
Артериальное давление
Наблюдалось значительное снижение среднего систолического АД при применении семаглутида 0,5 мг (3,5–5,1 мм рт.ст.) и 1 мг (5,4–7,3 мм рт.ст.) в комбинации с ПГГП или базальным инсулином. Не отмечалось значительной разницы по показателям диастолического АД между семаглутидом и препаратами сравнения.
Фармакокинетика
Период полувыведения (T1/2) семаглутида, равный приблизительно 1 неделе, делает возможным режим дозирования препарата Квинсента® 1 раз в неделю.
Абсорбция
Время достижения максимальной концентрации (Сmax) в плазме составило от 1 до 3 дней после введения дозы препарата.
Равновесная концентрация препарата (AUCτ/24) достигалась спустя 4–5 недель однократного еженедельного применения препарата. После п/к введения семаглутида в дозах 0,5 мг и 1 мг средние показатели его равновесной концентрации у пациентов с СД2 составили около 16 нмоль/л и 30 нмоль/л, соответственно.
Экспозиция для доз семаглутида 0,5 мг и 1 мг увеличивается пропорционально введенной дозе.
При п/к введении семаглутида в переднюю брюшную стенку, бедро или плечо достигается сходная экспозиция.
Абсолютная биодоступность семаглутида после п/к введения составила 89 %.
Распределение
Средний объем распределения семаглутида в тканях после п/к введения пациентам с СД2 составил приблизительно 12,5 л. Семаглутид в значительной степени связывался с альбумином плазмы крови (> 99 %).
Метаболизм
Семаглутид метаболизируется посредством протеолитического расщепления пептидной основы белка и последующего бета-окисления жирной кислоты боковой цепи.
Выведение
Желудочно-кишечный тракт (ЖКТ) и почки являются основными путями выведения семаглутида и его метаболитов. 2/3 введенной дозы семаглутида выводится почками, 1/3 – через кишечник.
Приблизительно 3 % от введенной дозы выводится почками в виде неизмененного семаглутида.
У пациентов с СД2 клиренс семаглутида составил около 0,05 л/ч. С элиминационным периодом полувыведения примерно 1 неделя семаглутид будет присутствовать в общем кровотоке в течение приблизительно 5 недель после введения последней дозы препарата.
Особые группы пациентов
Не требуется коррекции дозы семаглутида в зависимости от возраста, пола, расовой и этнической принадлежности, массы тела, наличия почечной или печеночной недостаточности.
Пациенты пожилого возраста
На основании данных, полученных в ходе КИ 3а фазы, включавших пациентов в возрасте от 20 до 86 лет, показано, что возраст не влиял на фармакокинетику семаглутида.
Пол
Пол не влиял на фармакокинетику семаглутида.
Раса
Расовая группа (европеоидная, негроидная или афроамериканская, азиатская) не влияла на фармакокинетику семаглутида.
Этническая принадлежность
Этническая принадлежность (латиноамериканская) не влияла на фармакокинетику семаглутида.
Масса тела
Масса тела влияла на экспозицию семаглутида. Более высокая масса тела приводит к более низкой экспозиции. Дозы семаглутида, равные 0,5 мг и 1 мг, обеспечивают достаточную экспозицию препарата в диапазоне массы тела от 40 до 198 кг.
Пациенты с почечной недостаточностью
Почечная недостаточность не оказывала клинически значимого эффекта на фармакокинетику семаглутида. Это было показано у пациентов с различной степенью почечной недостаточности (легкой, средней, тяжелой или у пациентов, находящихся на диализе) по сравнению с пациентами с нормальной функцией почек в исследовании однократной дозы семаглутида, равной 0,5 мг. Это также было показано на основании данных КИ 3а фазы для пациентов с СД2 и почечной недостаточностью, хотя опыт применения у пациентов с терминальной стадией заболевания почек был ограничен.
Пациенты с печеночной недостаточностью
Печеночная недостаточность не влияла на экспозицию семаглутида. Фармакокинетические свойства семаглутида оценивались в ходе исследования однократной дозы семаглутида, равной 0,5 мг у пациентов с различной степенью печеночной недостаточности (легкой, средней, тяжелой) по сравнению с пациентами с нормальной функцией печени.
Дети и подростки
Исследований семаглутида у детей и подростков до 18 лет не проводили.
Препарат Квинсента® показан для применения у взрослых пациентов с сахарным диабетом 2 типа на фоне диеты и физических упражнений для улучшения гликемического контроля в качестве:
монотерапии;
комбинированной терапии с другими пероральными гипогликемическими препаратами (ПГГП) – метформином, метформином и производным сульфонилмочевины, метформином и/или тиазолидиндионом у пациентов, не достигших адекватного гликемического контроля при проведении предшествующей терапии;
комбинированной терапии с инсулином у пациентов, не достигших адекватного гликемического контроля на терапии семаглутидом и метформином.
Препарат Квинсента® показан для снижения риска развития серьезных сердечно-сосудистых событий* у пациентов с сахарным диабетом 2 типа и высоким сердечно-сосудистым риском в качестве дополнения к стандартному лечению сердечно-сосудистых заболеваний (на основании анализа времени наступления первого серьезного сердечно-сосудистого события – см. раздел «Фармакологические свойства», подраздел «Оценка влияния на ССС»).
*серьезные сердечно-сосудистые события включают: смерть по причине сердечно-сосудистой патологии, инфаркт миокарда без смертельного исхода, инсульт без смертельного исхода.
Гиперчувствительность к семаглутиду или любому из вспомогательных веществ препарата
Медуллярный рак щитовидной железы в анамнезе, в том числе в семейном
Множественная эндокринная неоплазия (МЭН) 2 типа
Сахарный диабет 1 типа (СД1)
Диабетический кетоацидоз
Противопоказано применение препарата Квинсента® у следующих групп пациентов и при следующих состояниях/заболеваниях в связи с отсутствием данных по эффективности и безопасности или ограниченным опытом применения:
беременность и период грудного вскармливания;
возраст до 18 лет;
печеночная недостаточность тяжелой степени;
терминальная стадия почечной недостаточности (клиренс креатинина (КК) < 15 мл/мин);
хроническая сердечная недостаточность (ХСН) IV функционального класса (в соответствии с классификацией NYHA (Нью-Йоркская кардиологическая ассоциация)).
Семаглутид рекомендуется применять с осторожностью у пациентов с почечной недостаточностью и у пациентов с наличием панкреатита в анамнезе (см. раздел «Особые указания»).
Беременность
Исследования на животных продемонстрировали репродуктивную токсичность препарата (см. раздел «Особые указания», подраздел «Доклинические данные по безопасности»).
Данные по применению семаглутида у беременных женщин ограничены. Противопоказано применять семаглутид во время беременности. Женщинам с сохраненным репродуктивным потенциалом рекомендуется использовать контрацепцию во время терапии семаглутидом. Если пациентка готовится к беременности, либо беременность уже наступила, терапию семаглутидом необходимо прекратить. Из-за длительного периода полувыведения терапию семаглутидом необходимо прекратить как минимум за 2 месяца до планируемого наступления беременности (см. раздел «Фармакокинетика»).
Период грудного вскармливания
В исследованиях на животных у лактирующих крыс семаглутид проникал в молоко. Нельзя исключить риск для ребенка, находящегося на грудном вскармливании. Противопоказано применение семаглутида в период грудного вскармливания.
Режим дозирования
Начальная доза препарата Квинсента® составляет 0,25 мг 1 раз в неделю. После 4 недель применения дозу следует увеличить до 0,5 мг 1 раз в неделю. Для дальнейшего улучшения гликемического контроля после как минимум 4 недель применения препарата в дозе 0,5 мг 1 раз в неделю дозу можно увеличить до 1 мг 1 раз в неделю.
Доза препарата Квинсента® 0,25 мг не является терапевтической. Не рекомендуется введение более 1 мг в неделю.
Препарат Квинсента® может применяться в виде монотерапии или в комбинации с одним или более гипогликемическими препаратами (см. раздел «Клиническая эффективность и безопасность»).
При добавлении препарата Квинсента® к предшествующей терапии метформином и/или тиазолидиндионом, или ингибитором SGLT2 терапию метформином и/или тиазолидиндионом, или ингибитором SGLT2 можно продолжить в прежних дозах.
При добавлении препарата Квинсента® к проводимой терапии производным сульфонилмочевины или инсулином следует предусмотреть снижение дозы производного сульфонилмочевины или инсулина с целью снижения риска возникновения гипогликемии (см. раздел «Особые указания»).
Применение препарата Квинсента® не требует проведения самоконтроля концентрации глюкозы крови.
Самостоятельный мониторинг концентрации глюкозы в крови необходим для коррекции дозы сульфонилмочевины и инсулина, особенно в начале лечения препаратом Квинсента® и при снижении дозы инсулина. Рекомендуется использовать поэтапный подход к снижению дозы инсулина.
Пропущенная доза
В случае пропуска дозы препарат Квинсента® следует ввести как можно быстрее в течение 5 дней с момента запланированного введения дозы. Если продолжительность пропуска составляет более 5 дней, пропущенную дозу не нужно вводить. Следующую дозу препарата Квинсента® следует ввести в обычный запланированный день. В каждом случае пациенты могут возобновить их обычный однократный еженедельный график введения.
Особые группы пациентов
Пациенты пожилого возраста (≥ 65 лет)
Не требуется коррекции дозы в зависимости от возраста. Опыт применения семаглутида у пациентов в возрасте 75 лет и старше ограничен.
Пациенты с печеночной недостаточностью
Не требуется коррекции дозы у пациентов с печеночной недостаточностью (см. раздел «Фармакокинетика»). Опыт применения семаглутида у пациентов с печеночной недостаточностью тяжелой степени ограничен; применение препарата Квинсента® у таких пациентов противопоказано.
Пациенты с почечной недостаточностью
Не требуется коррекции дозы у пациентов с почечной недостаточностью. Опыт применения препарата у пациентов с терминальной стадией почечной недостаточности отсутствует; применение препарата Квинсента® у таких пациентов противопоказано.
Дети и подростки
Применение препарата Квинсента® у детей и подростков в возрасте до 18 лет противопоказано в связи с отсутствием данных по безопасности и эффективности.
Способ применения
Препарат Квинсента® применяют 1 раз в неделю в любое время, независимо от приема пищи.
Препарат Квинсента® вводят п/к в живот, бедро или плечо. Место инъекции может изменяться без коррекции дозы. Препарат Квинсента® нельзя вводить внутривенно и внутримышечно.
При необходимости день еженедельного введения можно менять при условии, что интервал времени между двумя инъекциями составляет не менее 3-х дней (> 72 часов). После выбора нового дня введения следует продолжить введение препарата 1 раз в неделю.
Руководство по применению
Препарат Квинсента® нельзя применять, если он выглядит иначе, чем прозрачный бесцветный или со слегка коричневатым оттенком раствор.
Препарат нельзя применять, если он подвергся замораживанию.
Предварительно заполненная мультидозовая одноразовая шприц-ручка для препарата Квинсента® поставляется в двух видах:
для препарата Квинсента®, раствор для подкожного введения, 0,25/0,5 мг/доза в предварительно заполненной шприц-ручке – позволяет вводить дозы 0,25 мг или 0,5 мг. Данная шприц-ручка предназначена для постепенного увеличения дозы от начальной –
0,25 мг и поддержания терапевтической дозы – 0,5 мг. Одна шприц-ручка содержит 1,5 мл раствора (2 мг семаглутида). Совместима с любыми иглами для шприц-ручек.
для препарата Квинсента®, раствор для подкожного введения, 0,25/0,5/1 мг/доза – в предварительно заполненной шприц-ручке позволяет вводить дозы 0,25 мг или 0,5 мг, или 1 мг. Одна шприц-ручка содержит 3 мл раствора (4 мг семаглутида). Совместима с любыми иглами для шприц-ручек.
Пациент должен быть проинформирован о том, что использованную иглу следует выбрасывать после каждой инъекции, а также о том, что нельзя хранить шприц-ручку с присоединенной иглой. Такая мера позволит предотвратить загрязнение, инфицирование и утечку препарата из шприц-ручки и гарантирует точность дозирования.
Шприц-ручка предназначена только для индивидуального использования.
Всегда после каждой инъекции удаляйте иглу и храните шприц-ручку с отсоединенной иглой. Это поможет предотвратить закупорку игл, загрязнение, заражение, вытекание раствора и введение неправильной дозы препарата.
Наиболее часто регистрируемыми нежелательными реакциями (HP) во время КИ являлись нарушения со стороны ЖКТ, включая тошноту, диарею и рвоту. В целом, данные реакции были легкой или средней степени тяжести и краткосрочными.
HP распределены по системно-органным классам в соответствии с MedDRA с указанием частоты их возникновения согласно рекомендациям ВОЗ: очень часто (≥ 1/10); часто (≥ 1/100 до < 1/10); нечасто (≥ 1/1000 до < 1/100); редко (≥ 1/10000 до < 1/1000); очень редко (< 1/10000) и неизвестно (невозможно оценить на основании имеющихся данных). В каждой группе частоты развития HP представлены по снижению степени серьезности.
Таблица 1. Hежелательные реакции, выявленные при проведении клинических исследований 3а фазы
|
Очень часто |
Часто |
Нечасто |
Редко |
Неизвестно |
|
Нарушения со стороны иммунной системы |
||||
|
Гиперчувствительностьс |
Анафилактические реакции |
|||
|
Нарушения метаболизма и питания |
||||
|
Гипогликемияа при совместном применении с инсулином или производным сульфонилмочевины |
Гипогликемияа при совместном применении с другими ПГГП |
|||
|
Снижение аппетита |
||||
|
Нарушения со стороны нервной системы |
||||
|
Головокружение |
Дисгевзия |
|||
|
Нарушения со стороны органа зрения |
||||
|
Осложнения диабетической ретинопатииb |
||||
|
Нарушения со стороны сердца |
||||
|
Увеличение частоты сердечных сокращений (ЧСС) |
||||
|
Желудочно-кишечные нарушения |
||||
|
Тошнота |
Рвота |
Острый панкреатит |
||
|
Диарея |
Боль в животе |
|||
|
Вздутие живота |
||||
|
Запор |
||||
|
Диспепсия |
||||
|
Гастрит |
||||
|
Гастроэзофагеальная рефлюксная болезнь |
||||
|
Отрыжка |
||||
|
Метеоризм |
||||
|
Нарушения со стороны печени и желчевыводящих путей |
||||
|
Холелитиаз |
||||
|
Нарушения со стороны кожи и подкожных тканей |
||||
|
Ангионевротический отекd |
||||
|
Общие нарушения и реакции в месте введения |
||||
|
Утомляемость |
Реакции в месте введения |
|||
|
Лабораторные и инструментальные данные |
||||
|
Повышение активности липазы |
||||
|
Повышение активности амилазы |
||||
|
Снижение массы тела |
||||
а Гипогликемия, определенная как тяжелая (требующая помощи другого человека) или симптоматическая в сочетании с концентрацией глюкозы в плазме крови < 3,1 ммоль/л.
b Осложнения диабетической ретинопатии – это сочетание из: необходимости в фотокоагуляции сетчатки глаза, необходимости в интравитреальном введении препаратов, кровоизлияния в стекловидное тело, развития слепоты, связанной с СД. Частота основана на исследовании СС исходов.
с Групповой термин, включающий также нежелательные реакции, связанные с гиперчувствительностью, такие как сыпь и крапивница.
d Нежелательные реакции из постмаркетинговых источников.
2-летнее исследование СС исходов и безопасности
В популяции пациентов с высоким риском развития СС заболеваний профиль HP был аналогичным таковому в других КИ 3а фазы (описаны в разделе «Клиническая эффективность и безопасность»).
Описание отдельных нежелательных реакций
Гипогликемия
Во время монотерапии семаглутидом не наблюдалось эпизодов тяжелой гипогликемии. Гипогликемия тяжелой степени, в основном, наблюдалась при применении семаглутида в комбинации с производным сульфонилмочевины или инсулином.
Наблюдалось несколько эпизодов тяжелой гипогликемии при применении семаглутида в комбинации с другими, за исключением производного сульфонилмочевины, ПГГП.
Гипогликемия по классификации Американской диабетической ассоциации наблюдалась у 11,3 % (0,3 случая/пациенто-год) пациентов при добавлении семаглутида в дозе 1,0 мг к терапии ингибитором SGLT2 по сравнению с 2,0 % (0,04 случая/пациенто-год) пациентов, получавших плацебо. О тяжелой гипогликемии сообщалось у 0,7 % (0,01 события/пациенто-год) и 0 % пациентов, соответственно.
НР со стороны ЖКТ
Во время терапии семаглутидом в дозах 0,5 мг и 1 мг у пациентов отмечалась тошнота, диарея и рвота. Большинство реакций были от легкой до средней степени тяжести и краткосрочными. НР стали причиной преждевременного выбывания из КИ 3,9 % и 5,9 % пациентов, соответственно. Чаще всего о нежелательных реакциях сообщалось в первые месяцы терапии.
Пациенты с низкой массой тела при лечении семаглутидом могут испытывать больше НР со стороны желудочно-кишечного тракта.
В КИ при одновременном применении ингибитора SGLT2 и семаглутида запор и гастроэзофагеальная рефлюксная болезнь наблюдались у 6,7 % и 4 % пациентов, получавших семаглутид 1,0 мг, соответственно, по сравнению с отсутствием событий у пациентов, получавших плацебо. Распространенность этих событий со временем не уменьшалась.
Нарушения со стороны печени и желчевыводящих путей
Сообщалось о возникновении холангита и холестатической желтухи.
Острый панкреатит
Частота развития острого панкреатита, подтвержденного по результатам экспертной оценки, в исследованиях 3а фазы составила 0,3 % при применении семаглутида и 0,2 % при применении препарата сравнения. В 2-летнем исследовании сердечно-сосудистых исходов частота развития острого панкреатита, подтвержденная по результатам экспертной оценки, составила 0,5 % при применении семаглутида и 0,6 % при применении плацебо (см. раздел «Особые указания»).
Осложнения диабетической ретинопатии
В 2-летнем КИ, в котором участвовали пациенты с СД2 и высоким СС риском, длительным течением СД и неадекватным контролем гликемии, подтвержденные случаи осложнений диабетической ретинопатии развивались у большего количества пациентов, получавших семаглутид (3,0 %), по сравнению с пациентами, получавшими плацебо (1,8 %). У пациентов с анамнезом диабетической ретинопатии в начале КИ возрастание абсолютного риска развития осложнений было выше. У пациентов с отсутствием подтвержденного анамнеза диабетической ретинопатии количество событий было одинаковым при применении семаглутида и плацебо.
В КИ продолжительностью до 1 года частота HP, связанных с диабетической ретинопатией, была одинаковой в группе семаглутида и препаратов сравнения.
Прекращение лечения по причине НР
Частота прекращения лечения по причине НР составила 8,7 % для пациентов, получавших семаглутид 1 мг. Наиболее частыми НР, приводившими к прекращению лечения, были нарушения со стороны ЖКТ.
Реакции в месте введения
Сообщалось о реакциях в месте введения (таких как сыпь в месте введения, покраснение) у 0,6 % и 0,5 % пациентов, получавших семаглутид 0,5 мг и 1 мг, соответственно. Эти реакции носили, как правило, легкий характер.
Иммуногенность
Вследствие потенциальных иммуногенных свойств белковых и пептидных лекарственных препаратов, у пациентов могут появиться антитела к семаглутиду после терапии. В конце КИ доля пациентов, у которых были обнаружены антитела к семаглутиду в любой момент времени, была низкой (1–2 %), и ни у одного пациента не было обнаружено нейтрализующих антител к семаглутиду или антител с нейтрализующим эндогенный ГПП-1 эффектом.
В ходе КИ сообщалось о передозировках до 4 мг в однократной дозе и до 4 мг в неделю. Наиболее частой НР, о которой сообщалось, была тошнота. Все пациенты выздоровели без осложнений.
Специфического антидота при передозировке препаратом Квинсента® не существует. В случае передозировки рекомендуется проведение соответствующей симптоматической терапии. Учитывая длительный период выведения препарата (примерно 1 неделя), может потребоваться продолжительный период наблюдения и лечения симптомов передозировки.
Исследования семаглутида in vitro показали очень небольшую вероятность ингибирования или индукции ферментов системы цитохрома Р450 (СYР) и ингибирования транспортеров лекарственных препаратов.
Задержка опорожнения желудка при применении семаглутида может оказывать влияние на всасывание сопутствующих пероральных лекарственных препаратов. Семаглутид следует применять с осторожностью у пациентов, получающих пероральные лекарственные препараты, для которых необходима быстрая абсорбция в ЖКТ.
Парацетамол
При оценке фармакокинетики парацетамола во время теста стандартизированного приема пищи было выявлено, что семаглутид задерживает опорожнение желудка. При одновременном применении семаглутида в дозе 1 мг AUC0-60 мин и Сmax парацетамола снизились на 27 % и 23 %, соответственно. Общая экспозиция парацетамола (AUC0-5 ч) при этом не изменялась. При одновременном приеме семаглутида и парацетамола коррекция дозы последнего не требуется.
Пероральные гормональные контрацептивные средства
Не предполагается, что семаглутид снижает эффективность пероральных гормональных контрацептивных средств. При одновременном применении комбинированного перорального гормонального контрацептивного препарата (0,03 мг этинилэстрадиола/0,15 мг левоноргестрела) и семаглутида последний не оказывал клинически значимого влияния на общую экспозицию этинилэстрадиола и левоноргестрела. Экспозиция этинилэстрадиола не была затронута; наблюдалось увеличение на 20 % экспозиции левоноргестрела в равновесном состоянии. Сmax не изменилась ни для одного из компонентов.
Аторвастатин
Семаглутид не изменял системную экспозицию аторвастатина после применения однократной дозы аторвастатина (40 мг). Сmax аторвастатина снизилась на 38 %. Это изменение было расценено как клинически незначимое.
Дигоксин
Семаглутид не изменял системную экспозицию или Сmax дигоксина после применения однократной дозы дигоксина (0,5 мг).
Метформин
Семаглутид не изменял системную экспозицию или Сmax метформина после применения метформина в дозе 500 мг 2 раза в день в течение 3,5 дней.
Варфарин
Семаглутид не изменял системную экспозицию или Сmax R- и S-изомеров варфарина после применения однократной дозы варфарина (25 мг). На основании определения международного нормализованного отношения (МНО) клинически значимых изменений фармакодинамических эффектов варфарина также не наблюдалось.
Несовместимость
Вещества, добавленные к препарату Квинсента®, могут вызвать деградацию семаглутида. Препарат Квинсента® нельзя смешивать с другими лекарственными средствами, в том числе с инфузионными растворами.
Применение препарата Квинсента® противопоказано у пациентов с СД1 или для лечения диабетического кетоацидоза.
Препарат Квинсента® не заменяет инсулин.
Диабетический кетоацидоз был зарегистрирован у инсулинозависимых пациентов, у которых отмечалось быстрое прекращение лечения или снижение дозы инсулина при начале лечения агонистом ГПП-1Р (см. раздел «Способ применения и дозы»).
Нежелательные реакции со стороны желудочно-кишечного тракта
Применение агонистов ГПП-1Р может быть ассоциировано с HP со стороны ЖКТ. Это следует учитывать при лечении пациентов с почечной недостаточностью, так как тошнота, рвота и диарея могут привести к дегидратации и ухудшению функции почек.
Острый панкреатит
При применении агонистов ГПП-1Р наблюдались случаи развития острого панкреатита. Пациенты должны быть проинформированы о характерных симптомах острого панкреатита. При подозрении на панкреатит терапия препаратом Квинсента® должна быть прекращена; в случае подтверждения острого панкреатита терапию препаратом Квинсента® возобновлять не следует. Следует соблюдать осторожность у пациентов с панкреатитом в анамнезе.
При отсутствии других признаков и симптомов острого панкреатита повышение активности ферментов поджелудочной железы не является прогностическим фактором развития острого панкреатита.
Гипогликемия
Пациенты, получающие препарат Квинсента® в комбинации с производным сульфонилмочевины или инсулином, могут иметь повышенный риск развития гипогликемии. В начале лечения препаратом Квинсента® риск развития гипогликемии можно снизить, уменьшив дозу производного сульфонилмочевины или инсулина.
Диабетическая ретинопатия
Наблюдалось повышение риска развития осложнений диабетической ретинопатии у пациентов с наличием диабетической ретинопатии, получающих терапию инсулином и семаглутидом (см. раздел «Побочное действие»). Следует соблюдать осторожность при применении семаглутида у пациентов с диабетической ретинопатией, получающих инсулинотерапию. Такие пациенты должны находиться под постоянным наблюдением и получать лечение в соответствии с клиническими рекомендациями. Быстрое улучшение гликемического контроля было ассоциировано с временным ухудшением состояния диабетической ретинопатии, однако при этом нельзя исключать и другие причины.
Сердечная недостаточность
Отсутствует опыт применения семаглутида у пациентов с ХСН IV функционального класса в соответствии с классификацией NYHA. Применение препарата у таких пациентов противопоказано.
Заболевания щитовидной железы
В пострегистрационном периоде применения другого аналога ГПП-1, лираглутида, были отмечены случаи медуллярного рака щитовидной железы (МРЩЖ). Имеющихся данных недостаточно для установления или исключения причинно-следственной связи возникновения МРЩЖ с применением аналогов ГПП-1. Необходимо проинформировать пациента о риске МРЩЖ и о симптомах опухоли щитовидной железы (появления уплотнения в области шеи, дисфагии, одышки, непроходящей охриплости голоса).
Значительное повышение концентрации кальцитонина в плазме крови может указывать на МРЩЖ (у пациентов с МРЩЖ значения концентрации кальцитонина в плазме крови обычно > 50 нг/л). При выявлении повышения концентрации кальцитонина в плазме крови следует провести дальнейшее обследование пациента. Пациенты с узлами в щитовидной железе, выявленными при медицинском осмотре или при проведении УЗИ щитовидной железы, также должны быть дополнительно обследованы.
Применение семаглутида у пациентов с личным или семейным анамнезом МРЩЖ или с синдромом МЭН типа 2 противопоказано.
Вспомогательные вещества
Препарат Квинсента® содержит менее 1 ммоль (23 мг) натрия на дозу 0,25/0,5 мг и 1 мг, то есть, по сути, не содержит натрия.
Доклинические данные по безопасности
Доклинические данные, основанные на исследованиях фармакологической безопасности, токсичности повторных доз и генотоксичности, не выявили какой-либо опасности для человека.
В 2-летних исследованиях канцерогенности у крыс и мышей в клинически значимых концентрациях семаглутид стал причиной развития опухолей
С-клеток щитовидной железы без смертельного исхода. Опухоли С-клеток щитовидной железы без смертельного исхода, наблюдаемые у крыс, характерны для группы аналогов ГПП-1. Считается, что в отношении людей данный риск является низким, но не может быть полностью исключен.
Фертильность
Действие семаглутида на фертильность у людей неизвестно. Семаглутид не влиял на фертильность самцов крыс. Среди самок крыс увеличение эстрального цикла и незначительное снижение количества овуляций наблюдалось при дозах, сопровождавшихся снижением массы тела самки.
Семаглутид не влияет или незначительно влияет на способность управлять транспортными средствами или работу с механизмами. Пациенты должны быть предупреждены о том, что им следует соблюдать меры предосторожности во избежание развития у них гипогликемии во время управления транспортными средствами и при работе с механизмами, особенно при применении препарата Квинсента® в комбинации с производным сульфонилмочевины или инсулином.
Раствор для подкожного введения, 0,25/0,5 мг/доза, 0,25/0,5/1 мг/доза.
По 1,5 мл препарата, содержащего 2 мг семаглутида (для дозировки 0,25/0,5 мг/доза) или 3 мл препарата, содержащего 4 мг семаглутида (для дозировки 0,25/0,5/1 мг/доза) в картриджи из стекла I гидролитического класса для лекарственных средств с плунжерами резиновыми, укупоренные обкатанными комбинированными алюминиевыми колпачками с резиновой прокладкой для картриджей.
Картридж установлен в пластиковую мультидозовую одноразовую шприц-ручку для многократных инъекций.
1, 2 или 3 пластиковые мультидозовые одноразовые шприц-ручки для многократных инъекций в контурной ячейковой упаковке или без контурной ячейковой упаковки, вместе с инструкцией по медицинскому применению помещают в картонную пачку с контролем первого вскрытия или без него.
Дополнительно картонная пачка с пластиковыми мультидозовыми одноразовыми шприц-ручками для многократных инъекций может комплектоваться стерильными одноразовыми иглами в количестве:
– 6 штук (для дозировки 0,25/0,5 мг/доза), 4, 6, 9 штук (для дозировки 0,25/0,5/1 мг/доза) для 1 шприц-ручки;
– 12 штук (для дозировки 0,25/0,5 мг/доза), 8, 12, 18 штук (для дозировки 0,25/0,5/1 мг/доза) для 2 шприц-ручек;
– 18 штук (для дозировки 0,25/0,5 мг/доза), 12, 18, 27 штук (для дозировки 0,25/0,5/1 мг/доза) для 3 шприц-ручек
Хранить при температуре от 2 до 8 °С (в холодильнике), но не рядом с морозильной камерой. Защищать от света. Не замораживать.
Используемую или переносимую в качестве запасной шприц-ручку с препаратом хранить при температуре не выше 30 °С или при температуре от 2 до 8 °С (в холодильнике) в течение 6 недель. Не замораживать.
После использования закрывать шприц-ручку колпачком для защиты от света.
ООО «ПРОМОМЕД РУС», Россия
129090, г. Москва, проспект Мира, дом 13, строение 1, офис 13
АО «Биохимик», Россия
Юридический адрес:
430030, Республика Мордовия, г. Саранск, ул. Васенко, д. 15А
Адрес производства:
430030, Республика Мордовия, г. Саранск, ул. Васенко, д. 15А
Телефон: +7 (8342) 38-03-68
Адрес электронной почты: biohimic@promomed.pro
Адрес в сети интернет: www.promomed.ru
ООО «Завод Медсинтез», Россия
Юридический адрес:
620028, Свердловская обл., г. Екатеринбург, ул. Кирова, стр. 28, пом. 205
Адрес производства:
624130, Свердловская обл., г. Новоуральск, ул. Торговая, зд. 15, стр. 3
Тел.: +7 (34370) 2-50-61
Факс: +7 (34370) 2-54-95
Организация, принимающая претензии потребителей
ООО «ПРОМОМЕД РУС», Россия
129090, г. Москва, проспект Мира, дом 13, строение 1, офис 13
Телефон: +7 (495) 640-25-28
Круглосуточный телефон горячей линии фармаконадзора: 8-800-777-86-04 (бесплатно)
Адрес электронной почты: reception@promomed.pro
В 1 мл препарата содержится:
Действующее вещество: лираглутид 6,0 мг (в одной предварительно заполненной мультидозовой одноразовой шприц-ручке объемом 3 мл содержится 18 мг лираглутида)
Вспомогательные вещества: натрия гидрофосфата дигидрат, фенол, пропиленгликоль, 2 М раствор натрия гидроксида или 2 М раствор хлористоводородной кислоты (для коррекции рН), вода для инъекций.
Прозрачная бесцветная или коричневатого цвета жидкость
Гипогликемическое средство – аналог глюкагоноподобного пептида-1 (ГПП-1)
А10ВJ02
Фармакодинамика
Действующее вещество препарата Энлигрия® - лираглутид - это химически синтезированный пептид, который представляет собой ацилированный аналог человеческого ГПП-1, имеющий 97 % гомологий с аминокислотной последовательностью эндогенного человеческого ГПП-1.
Лираглутид связывается и активирует рецептор ГПП-1 (ГПП-1Р). Лираглутид устойчив к метаболическому распаду, период его полувыведения из плазмы после подкожного введения составляет 13 ч.
Длительный период полувыведения препарата из плазмы обеспечивается тремя механизмами: самоассоциацией, в результате которой происходит замедленное всасывание препарата, связыванием с альбумином и более высоким уровнем ферментативной стабильности по отношению к дипептидилпептидазе-4 (ДПП-4) и ферменту нейтральной эндопептидазы (НЭП).
ГПП-1 является физиологическим регулятором аппетита и потребления пищи, а ГПП-1Р расположены в нескольких областях головного мозга, участвующих в процессах регуляции аппетита.
В исследованиях на животных периферическое введение лираглутида приводило к связыванию с рецепторами в специфических областях головного мозга, включая гипоталамус, где лираглутид посредством специфической активации ГПП-1Р усиливал сигналы насыщения и ослаблял сигналы голода, тем самым приводя к уменьшению массы тела.
ГПП-1Р представлены также в специфических областях сердца, сосудов, иммунной системы и почек. В экспериментах на мышах с атеросклерозом лираглутид предупреждал дальнейшее развитие аортальных бляшек и снижал в них воспаление. В дополнение, лираглутид оказывал благоприятный эффект на липиды в плазме. Лираглутид не уменьшал размер уже существующих бляшек.
Лираглутид уменьшает массу тела у человека преимущественно посредством уменьшения массы жировой ткани. Уменьшение массы тела происходит за счет уменьшения потребления пищи. Лираглутид не увеличивает 24-часовой расход энергии. Лираглутид регулирует аппетит при помощи усиления чувства наполнения желудка и насыщения, одновременно ослабляя чувство голода и уменьшая предполагаемое потребление пищи.
Лираглутид стимулирует секрецию инсулина и уменьшает неоправданно высокую секрецию глюкагона глюкозозависимым образом, а также улучшает функцию бета-клеток поджелудочной железы, что приводит к снижению концентрации глюкозы натощак и после приема пищи. Механизм снижения концентрации глюкозы также включает небольшую задержку опорожнения желудка.
В долгосрочных клинических исследованиях (КИ) с участием пациентов с избыточной массой тела и ожирением применение лираглутида в сочетании с низкокалорийной диетой и усиленной физической активностью приводило к значительному снижению массы тела.
Влияние на аппетит, потребление калорий и расход энергии, опорожнение желудка и концентрацию глюкозы натощак и после приема пищи Фармакодинамические эффекты лираглутида изучались в пятинедельном фармакологическом КИ с участием 49 пациентов с ожирением (индекс массы тела (ИМТ) 30–40 кг/м2) без сахарного диабета (СД).
Аппетит, потребление калорий и расход энергии
Считается, что снижение массы тела при применении лираглутида связано с регулированием аппетита и количества потребляемой пищи. Аппетит оценивали перед и в течение 5 ч после стандартизированного завтрака; неограниченное потребление пищи оценивали во время последующего обеда. По сравнению с плацебо лираглутид увеличивал чувство насыщения и наполнения желудка после приема пищи и уменьшал чувство голода и оценочное количество предполагаемого потребления пищи, а также уменьшал неограниченное потребление пищи. При оценке с помощью респираторной камеры не было отмечено связанного с терапией увеличения 24-часового расхода энергии.
Опорожнение желудка
Применение лираглутида приводило к небольшой задержке опорожнения желудка в течение первого часа после приема пищи, в результате чего уменьшалась скорость повышения концентрации глюкозы, а также общая концентрация глюкозы крови после приема пищи.
Концентрации глюкозы, инсулина и глюкагона натощак и после приема пищи
Концентрации глюкозы, инсулина и глюкагона натощак и после приема пищи оценивали перед и в течение 5 ч после стандартизированного приема пищи. По сравнению с плацебо лираглутид уменьшал концентрацию глюкозы натощак и после приема пищи (AUC0-60 мин) в течение первого часа после приема пищи, а также уменьшал 5-часовую площадь под кривой «концентрация-время» (AUC) глюкозы и нарастающую концентрацию глюкозы (AUC0-300 мин). Кроме того, по сравнению с плацебо лираглутид уменьшал постпрандиальные концентрации глюкагона (AUC0-300 мин) и инсулина (AUC0-60 мин) и нарастающую концентрацию инсулина (iAUC0-60 мин) после приема пищи.
Концентрации глюкозы и инсулина натощак, и нарастающие концентрации глюкозы и инсулина также оценивали во время перорального теста толерантности к глюкозе (ПТТГ) с 75 г глюкозы до начала терапии и через 1 год терапии у 3731 пациента с избыточной массой тела и с ожирением, а также с наличием или отсутствием предиабета. По сравнению с плацебо лираглутид уменьшал концентрацию натощак и нарастающую концентрацию глюкозы. Эффект был более выраженным у пациентов с предиабетом. Кроме того, по сравнению с плацебо лираглутид уменьшал концентрацию инсулина натощак и увеличивал нарастающую концентрацию инсулина.
После 160 недель продолжающейся терапии лираглутидом 3,0 мг AUC глюкозы плазмы снизилась, в то время как при применении плацебо оставалась неизменной. Дополнительно AUC инсулина оставалась относительно стабильной в течение 160-недельного периода лечения лираглутидом 3,0 мг, в то время как при применении плацебо наблюдалось ее снижение. Все изученные эффекты от проводимой терапии были статистически значимыми в пользу лираглутида 3,0 мг.
Влияние на концентрацию глюкозы натощак и нарастающую концентрацию глюкозы у пациентов с сахарным диабетом 2 (СД2) типа с избыточной массой тела или ожирением
По сравнению с плацебо лираглутид снижал концентрацию глюкозы натощак и среднюю нарастающую постпрандиальную концентрацию глюкозы (через 90 минут после приема пищи, среднее значение для 3-х приемов пищи в сутки).
Функция бета-клеток поджелудочной железы
КИ продолжительностью до одного года с применением лираглутида у пациентов с избыточной массой тела, а также с наличием или отсутствием СД продемонстрировали улучшение и сохранение функции бета-клеток поджелудочной железы. Это было показано с использованием таких методов измерения, как гомеостатическая модель оценки функции бета-клеток (НОМА-В) и соотношение концентраций проинсулина и инсулина.
Клиническая эффективность и безопасность
Эффективность и безопасность применения лираглутида для длительной коррекции массы тела в сочетании с низкокалорийной диетой и усилением физической активности была изучена в 4-х рандомизированных двойных слепых плацебо-контролируемых исследованиях 3 фазы SCALE, включавших в общей сложности 5358 взрослых пациентов.
Масса тела
По сравнению с плацебо при применении лираглутида было достигнуто более выраженное снижение массы тела у пациентов с ожирением/избыточной массой тела во всех исследованных группах, в том числе с наличием или отсутствием предиабета, СД2 и обструктивным апноэ во сне средней или тяжелой степени. Кроме того, среди популяции исследования большая часть пациентов достигла снижения массы тела ≥ 5 % и > 10 % при применении лираглутида по сравнению с плацебо. Значительное снижение массы тела наблюдалось также в КИ, в котором пациенты достигли среднего показателя снижения массы тела 6,0 % с помощью низкокалорийной диеты в течение 12 недель до начала лечения лираглутидом. В этом исследовании большее количество пациентов сохранили потерю массы тела, которая была достигнута до начала лечения лираглутидом по сравнению с плацебо (81,4 % и 48,9 %, соответственно).
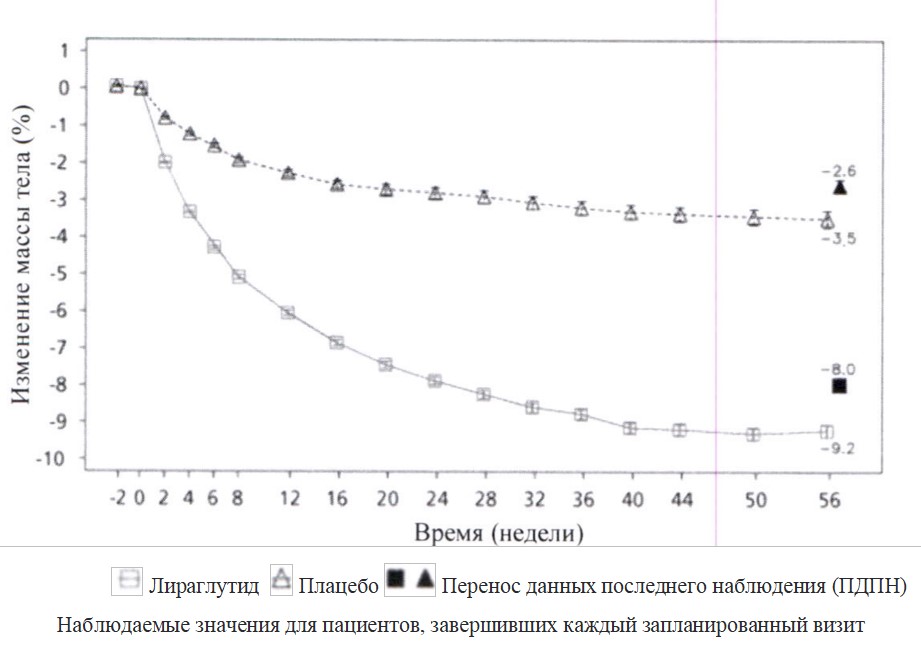
Рисунок 1. Изменение массы тела (%) в динамике по сравнению с исходным значением у пациентов (0-56 недели).
В КИ длительностью 160 недель пациенты, получавшие лираглутид, достигли более значительной потери массы тела по сравнению с пациентами, получавшими плацебо. В основном потеря массы тела произошла в первый год и удерживалась на протяжении 160 недель.
В КИ длительностью 160 недель средний процент изменения массы тела и доля пациентов, достигших потери массы тела от исходного значения до 160 недели не менее чем на 5 % и более 10 %, были также значительными по сравнению с плацебо.
Снижение массы тела после 12 недель терапии лираглутидом (лираглутид 3,0 мг)
В двух исследованиях продолжительностью 56 недель после 12 недель терапии лираглутидом в дозе 3,0 мг 67,5 % и 50,4 % пациентов достигли снижения массы тела не менее чем на 5 %. Для этих пациентов среднее снижение массы тела на момент завершения исследования (1 год) составило 11,2 % по сравнению с исходным значением. У пациентов, достигших снижения массы тела менее чем на 5 % после 12 недель терапии в дозе 3,0 мг и завершивших исследование (1 год), среднее снижение массы тела составило 3,8 %.
Контроль гликемии
Терапия лираглутидом существенно улучшала гликемические показатели в субпопуляциях с нормогликемией, предиабетом и СД2.
Через 56 недель у 0,2 % пациентов, получавших лираглутид, по сравнению с 1,1 % в группе плацебо развился СД2. У 69,2 % пациентов с предиабетом наблюдалось обратное развитие этого состояния при применении лираглутида по сравнению с 32,7 % в группе плацебо.
На 160-й неделе лечения диагноз СД2 был поставлен 3 % пациентов, получавшим лираглутид, и 11 % пациентов, получавшим плацебо. По сравнению с плацебо время до развития СД2 при применении лираглутида 3,0 мг было в 2,7 раза больше, а относительный риск (ОР) развития СД2 при применении лираглутида равен 0,2. На 160-й неделе в группе лираглутида 3,0 мг у 65,9 % пациентов с предиабетом наблюдалось обратное развитие этого состояния до нормогликемии по сравнению с 36,3 % в группе плацебо.
В одном из исследований 69,2 % и 56,5 % пациентов с ожирением и СД2, получавших лираглутид, достигли целевого значения НbА1С < 7 % и ≤ 6,5 %, соответственно, по сравнению с 27,2 % и 15,0 % у пациентов, получавших плацебо.
Кардиометаболические параметры
По сравнению с плацебо лираглутид значительно улучшал показатели систолического артериального давления (АД), окружности талии и концентрации липидов натощак.
В КИ длительностью 160 недель среднее уменьшение окружности талии составило 8,2 см при применении лираглутида и 4,0 см при применении плацебо; уменьшение показателей систолического и диастолического АД составило 4,3 мм рт. ст. и 1,5 мм рт. ст. при применении лираглутида и 2,7 мм рт. ст. и 1,8 мм рт. ст. при применении плацебо соответственно; уменьшение концентрации холестерина липопротеинов низкой плотности (ЛПНП) составило 3,1 ммоль/л при применении лираглутида и 0,7 ммоль/л при применении плацебо; увеличение концентрации холестерина липопротеинов высокой плотности (ЛПВП) составило 2,3 ммоль/л при применении лираглутида и 0,5 ммоль/л при применении плацебо.
Индекс апноэ-гипноэ (ИАГ)
По сравнению с плацебо при применении лираглутида наблюдалось существенное снижение тяжести обструктивного апноэ во сне, которая оценивалась по изменению ИАГ относительно исходного значения.
Иммуногенность
Вследствие потенциальных иммуногенных свойств белковых и пептидных лекарственных препаратов, у пациентов могут появиться антитела к лираглутиду после терапии. В КИ у 2,5 % пациентов, получавших лираглутид, появились антитела к лираглутиду. Образование антител не привело к снижению эффективности препарата.
Оценка сердечно-сосудистых событий
Серьезные сердечно-сосудистые события (СССС) были оценены группой внешних независимых экспертов и определены как инфаркт миокарда без смертельного исхода, инсульт без смертельного исхода и смерть по причине сердечно-сосудистой патологии. В 5 двойных слепых контролируемых КИ 2 и 3 фазы с применением лираглутида было отмечено 6 СССС у пациентов, получавших лираглутид и 10 СССС – у получавших плацебо пациентов. ОР с 95 % доверительным интервалом (ДИ) составил 0,33 при применении лираглутида по сравнению с плацебо. В КИ 3 фазы среднее увеличение частоты сердечных сокращений (ЧСС) составило 2,5 удара в минуту у пациентов, получавших лираглутид. Наибольшее увеличение ЧСС наблюдалось после 6 недель терапии. Это увеличение было обратимым и исчезало после прекращения терапии лираглутидом.
Было проведено многоцентровое, плацебо-контролируемое, двойное слепое КИ «Эффект и воздействие лираглутида при сахарном диабете: оценка сердечно-сосудистых рисков» (LEADER®).
По сравнению с плацебо лираглутид 1,8 мг существенно снижал риск развития СССС (Рисунок 2). ОР развития СССС был стабильно ниже 1 для всех трех сердечно-сосудистых событий.
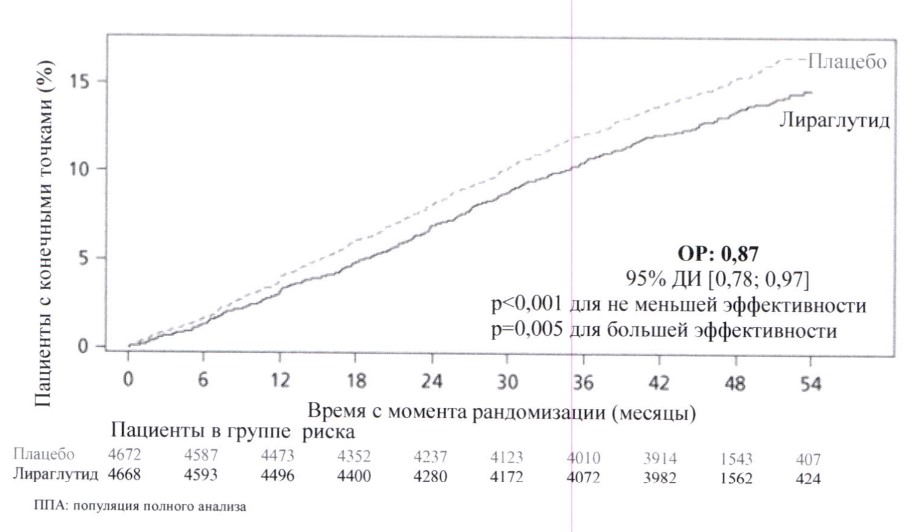
Рисунок 2. График Каплан-Мейера - время до возникновения первого СССС - Популяция полного анализа (ППА).
Лираглутид 1,8 мг также существенно снижал риск развития расширенных СССС (первичные СССС, нестабильная стенокардия, приводящая к госпитализации, реваскуляризация миокарда или госпитализация по причине сердечной недостаточности) и прочих вторичных конечных точек.
Дети и подростки
Европейское агентство лекарственных средств отложило обязательство по представлению результатов исследований лираглутида в одной или нескольких группах детей для лечения ожирения (см. раздел «Способ применения и дозы»).
В двойном слепом КИ, сравнивающем эффективность и безопасность лираглутида относительно плацебо в отношении снижения массы тела у подростков в возрасте 12 лет и старше с ожирением, лираглутид превосходил плацебо в снижении стандартного отклонения ИМТ (измеренного для оценки снижения массы тела) после 56 недель лечения.
Большее количество пациентов достигло ≥ 5 % и ≥ 10 % снижения ИМТ на терапии лираглутидом, чем пациенты, получавшие плацебо, при более значительном снижении среднего ИМТ и массы тела. В течение 26 недель последующего наблюдения без применения препарата наблюдалось восстановление массы тела в группе терапии лираглутидом по сравнению с плацебо (оценивалось как изменение стандартного отклонения ИМТ).
Исходя из переносимости лираглутида, большинству пациентов (82,4 %) доза препарата была увеличена до 3,0 мг, и они продолжили получать эту дозу, остальным пациентам дозы были увеличены, в соответствии с переносимостью терапии, и они продолжили получать препарат в диапазоне доз 0,6 мг - 2,4 мг (2,4 % – дозу 0,6 мг, 3,2 % – дозу 1,2 мг, 3,2 % – дозу 1,8 мг, 8,8 % – дозу 2,4 мг).
После 56 недель терапии препаратом не было выявлено влияния на рост или половое развитие.
Было проведено исследование с 16-недельным двойным слепым периодом и 36-недельным открытым периодом для оценки эффективности и безопасности лираглутида у детей и подростков с синдромом Прадера-Вилли и ожирением. В исследование были включены 32 пациента в возрасте от 12 до < 18 лет (часть А) и 24 пациента в возрасте от 6 до < 12 лет (часть В). Пациенты были рандомизированы в соотношении 2:1 для получения терапии лираглутидом или плацебо. У пациентов с массой тела менее 45 кг увеличение дозы начинали с более низкой дозы; 0,3 мг вместо 0,6 мг с повышением до максимальной дозы 2,4 мг.
Установленная разница средней оценки стандартного отклонения ИМТ (SDS ИМТ) после 16 недель (часть А: -0,20 против -0,13, часть В: -0,50 против -0,44) и после 52 недель (часть А: -0,31 против -0,17, часть В: -0,73 против -0,67) была сопоставимой на терапии лираглутидом и плацебо.
В ходе исследований не было выявлено никаких дополнительных рисков в отношении безопасности препарата.
Результаты определяемых пациентами показателей
Лираглутид по сравнению с плацебо улучшал определяемые пациентами оценки по нескольким показателям. Было отмечено значительное улучшение общей оценки по Упрощенному опроснику влияния массы тела на качество жизни (IWQoL-Lite) и по всем шкалам опросника для оценки качества жизни SF-36, что указывает на положительное влияние на физические и психологические параметры качества жизни.
Фармакокинетика
Всасывание
Всасывание лираглутида после подкожного введения происходит медленно, время достижения максимальной концентрации (tmах) – около 11 ч после введения. У пациентов с ожирением (ИМТ 30–40 кг/м2) после введения лираглутида в дозе 3,0 мг средняя равновесная концентрация лираглутида (AUCτ/24) достигает приблизительно 31 нмоль/л. В диапазоне доз от 0,6 мг до 3,0 мг экспозиция лираглутида увеличивается пропорционально дозе. Абсолютная биодоступность лираглутида после подкожного введения составляет приблизительно 55 %.
Распределение
Средний кажущийся объем распределения после подкожного введения лираглутида в дозе 3,0 мг составляет 20–25 л (у лиц с массой тела около 100 кг). Лираглутид в значительной степени связывается с белками плазмы крови (> 98 %).
Метаболизм
На протяжении 24 ч после введения здоровым добровольцам однократной дозы [3Н]-лираглутида главным компонентом в плазме оставался неизмененный лираглутид. Были обнаружены 2 метаболита (≤ 9 % и ≤ 5 % от уровня общей радиоактивности в плазме крови).
Выведение
Лираглутид метаболизируется эндогенно без участия какого-либо специфического органа в качестве основного пути выведения. После введения дозы [3Н]-лираглутида неизмененный лираглутид не определялся в моче или кале. Лишь незначительная часть введенной радиоактивности в виде метаболитов лираглутида выводилась почками или через кишечник (6 % и 5 % соответственно). Радиоактивные вещества выделяются почками или через кишечник, в основном в течение первых 6–8 дней и представляют собой 3 метаболита.
Средний клиренс после подкожного введения 3,0 мг лираглутида составляет приблизительно 0,9–1,4 л/ч, период полувыведения составляет примерно 13 ч.
Особые группы пациентов
Пациенты пожилого возраста
Коррекции дозы с учетом возраста не требуется. Согласно результатам популяционного фармакокинетического анализа у пациентов с избыточной массой тела и ожирением в возрасте 18–82 лет возраст не оказывал клинически значимого влияния на фармакокинетику лираглутида при подкожном введении в дозе 3,0 мг.
Пол
Основываясь на данных популяционного фармакокинетического анализа, у женщин скорректированный по массе тела клиренс лираглутида после подкожного введения в дозе 3,0 мг на 24 % меньше, чем у мужчин. На основании данных по ответной реакции на воздействие препарата, коррекции дозы с учетом пола не требуется.
Этническая принадлежность
Согласно результатам популяционного фармакокинетического анализа, в который были включены данные исследований у пациентов с избыточной массой тела и ожирением европеоидной, негроидной, азиатской и латиноамериканской расовых групп, этническая принадлежность не оказывала клинически значимого влияния на фармакокинетику лираглутида при подкожном введении в дозе 3,0 мг.
Масса тела
Экспозиция лираглутида уменьшается при увеличении исходной массы тела. Применение лираглутида в дозе 3,0 мг ежедневно обеспечивает адекватную экспозицию в диапазоне массы тела 60–234 кг, согласно оценке ответной реакции на системную экспозицию препарата в КИ.
Экспозицию лираглутида у пациентов с массой тела больше 234 кг не изучали.
Пациенты с печеночной недостаточностью
Фармакокинетику лираглутида оценивали у пациентов с различной степенью нарушения функции печени в исследовании однократной дозы (0,75 мг). Наблюдалось снижение экспозиции лираглутида на 13–23 % у пациентов с печеночной недостаточностью легкой и средней степени тяжести и значительное снижение экспозиции лираглутида (на 44 %) у пациентов с печеночной недостаточностью тяжелой степени (> 9 баллов по классификации Чайлд-Пью) в сравнении со здоровыми добровольцами.
Пациенты с почечной недостаточностью
В исследовании однократной дозы (0,75 мг) экспозиция лираглутида была меньше у пациентов с почечной недостаточностью по сравнению с лицами с нормальной функцией почек.
Экспозиция лираглутида была меньше на 33 %, 14 %, 27 % и 26 %, соответственно, у пациентов с почечной недостаточностью легкой (клиренс креатинина (КК) 50–80 мл/мин), средней (КК 30–50 мл/мин), тяжелой степени (КК < 30 мл/мин) и у пациентов с терминальной стадией почечной недостаточности, нуждающихся в гемодиализе.
Дети и подростки
Фармакокинетические свойства лираглутида 3,0 мг оценивались в клинических исследованиях у подростков с ожирением в возрасте от 12 до 18 лет (134 пациента, масса тела 62–178 кг).
Экспозиция лираглутида у подростков в возрасте от 12 до 18 лет была сопоставима с наблюдаемой у взрослых пациентов с ожирением.
Фармакокинетические свойства были также оценены в клиническом фармакологическом исследовании с участием детей с ожирением в возрасте от 7 до 11 лет (13 пациентов, масса тела 54–87 кг). Экспозиция лираглутида 3,0 мг у детей в возрасте от 7 до 11 лет была сопоставима с наблюдаемой у взрослых пациентов после поправки на массу тела.
Доклинические данные по безопасности
Доклинические данные, основанные на исследованиях фармакологической безопасности, токсичности повторных доз и генотоксичности, не выявили какой-либо опасности для человека.
В двухлетних исследованиях канцерогенности у крыс и мышей были выявлены опухоли С-клеток щитовидной железы, не приводившие к летальному исходу. Результаты, полученные в ходе исследований на грызунах, обусловлены тем, что грызуны проявляют особую чувствительность в отношении опосредуемого рецептором ГПП-1 негенотоксичного специфического механизма. Появления других новообразований, связанных с проводимой терапией, отмечено не было.
В исследованиях на животных не выявлено прямого неблагоприятного эффекта препарата на фертильность, но было отмечено незначительное увеличение частоты ранней эмбриональной смерти при применении самых высоких доз препарата.
У молодых крыс лираглутид вызывал задержку полового созревания как у самцов, так и у самок при клинически значимом воздействии. Эти задержки не повлияли на фертильность и репродуктивную способность обоих полов или на способность самок сохранять беременность.
Взрослые
Препарат Энлигрия® показан в качестве дополнения к низкокалорийной диете и усиленной физической нагрузке для длительного применения с целью коррекции массы тела у взрослых пациентов с ИМТ:
Подростки
Препарат Энлигрия® может быть использован в качестве дополнения к здоровому питанию и усиленной физической нагрузке с целью коррекции массы тела у подростков в возрасте от 12 лет и старше с:
Таблица 1. *Пороговое значение ИМТ IOTF для ожирения в зависимости от пола в возрасте от 12 до 18 лет
|
Возраст (количество лет) |
Индекс массы тела 30 кг/м2 |
|
|
Мужской пол |
Женский пол |
|
|
12 |
26,02 |
26,67 |
|
12,5 |
26,43 |
27,24 |
|
13 |
26,84 |
27,76 |
|
13,5 |
27,25 |
28,20 |
|
14 |
27,63 |
28,57 |
|
14,5 |
27,98 |
28,87 |
|
15 |
28,30 |
29,11 |
|
15,5 |
28,60 |
29,29 |
|
16 |
28,88 |
29,43 |
|
16,5 |
29,14 |
29,56 |
|
17 |
29,41 |
29,69 |
|
17,5 |
29,70 |
29,84 |
|
18 |
30,00 |
30,00 |
Гиперчувствительность к лираглутиду или любому из вспомогательных веществ препарата
Медуллярный рак щитовидной железы в анамнезе, в том числе в семейном
Множественная эндокринная неоплазия 2 типа
Тяжелая депрессия, суицидальные мысли или поведение, в том числе в анамнезе.
Противопоказано применение у следующих групп пациентов и при следующих состояниях/заболеваниях в связи с отсутствием данных по эффективности и безопасности:
почечная недостаточность тяжелой степени (КК < 30 мл/мин);
печеночная недостаточность тяжелой степени;
детский возраст до 12 лет;
подростки в возрасте от 12 до 18 лет с массой тела ≤ 60 кг;
у пациентов в возрасте ≥ 75 лет;
период беременности и грудного вскармливания;
хроническая сердечная недостаточность (ХСН) IV функционального класса (в соответствии с классификацией NYHA (Нью-Йоркская кардиологическая ассоциация));
одновременное применение других препаратов для коррекции массы тела;
применение в комбинации с другими агонистами рецепторов ГПП-1;
вторичное ожирение на фоне эндокринологических заболеваний или расстройств пищевого поведения или на фоне применения лекарственных препаратов, которые могут привести к увеличению массы тела.
У пациентов с сахарным диабетом лираглутид не должен применяться в качестве заменителя инсулина.
Опыт применения лираглутида у пациентов с воспалительными заболеваниями кишечника и диабетическим парезом желудка ограничен. Применение лираглутида у таких пациентов не рекомендуется, поскольку оно связано с транзиторными нежелательными реакциями со стороны желудочно-кишечного тракта (ЖКТ), включая тошноту, рвоту и диарею.
Лираглутид рекомендуется применять с осторожностью у пациентов с печеночной недостаточностью легкой и средней степени тяжести, заболеваниями щитовидной железы и наличием острого панкреатита в анамнезе.
Беременность
Данные по применению лираглутида у беременных женщин ограничены.
В исследованиях на животных была продемонстрирована репродуктивная токсичность.
Потенциальный риск для человека неизвестен.
Применение препарата Энлигрия® в период беременности противопоказано. При планировании или наступлении беременности терапию препаратом Энлигрия® необходимо прекратить.
Период грудного вскармливания
Неизвестно, проникает ли лираглутид в грудное молоко человека. В исследованиях на животных было продемонстрировано, что проникновение лираглутида и структурно близких метаболитов в грудное молоко является низким. В доклинических исследованиях было продемонстрировано связанное с терапией замедление роста новорожденных крысят, находящихся на грудном вскармливании. В связи с отсутствием опыта применения, препарат Энлигрия® противопоказан во время грудного вскармливания.
Фертильность
За исключением незначительного уменьшения числа живых эмбрионов, результаты исследований на животных не указывают на наличие неблагоприятного влияния на фертильность (см. подраздел «Доклинические данные по безопасности»).
Дозы
Взрослые
Начальная доза составляет 0,6 мг в сутки. Дозу увеличивают до 3,0 мг в сутки, прибавляя по 0,6 мг с интервалами не менее одной недели для улучшения желудочно-кишечной переносимости (см. Таблицу 2). Если при увеличении дозы новая доза плохо переносится пациентом в течение 2 недель подряд, следует рассмотреть вопрос о прекращении терапии.
Применение препарата в суточной дозе больше 3,0 мг не рекомендуется.
Терапию препаратом Энлигрия® следует прекратить, если после 12 недель применения препарата в дозе 3,0 мг в сутки потеря в массе тела составила менее 5 % от исходного значения.
Таблица 2. Схема увеличения дозы
|
Доза |
Недели |
|
|
Увеличение дозы в течение 4 недель |
0,6 мг |
1 |
|
1,2 мг |
1 |
|
|
1,8 мг |
1 |
|
|
2,4 мг |
1 |
|
|
Терапевтическая доза |
3,0 мг |
|
Подростки (≥ 12 лет)
Для подростков в возрасте от 12 до 18 лет следует применять такой же режим увеличения дозы, как и для взрослых (см. Таблицу 2). Дозу препарата следует увеличивать до тех пор, пока не будет достигнуто значение 3,0 мг (терапевтическая доза) или максимально переносимая доза.
Применение препарата в суточной дозе больше 3,0 мг не рекомендуется.
Терапию препаратом Энлигрия® следует прекратить и пересмотреть, если после 12 недель применения препарата в дозе 3,0 мг в сутки или максимальной переносимой дозе пациенты потеряли менее 4 % от своего ИМТ или z-показателя ИМТ.
Пропущенная доза
Если после обычного времени введения дозы прошло менее 12 ч, пациент должен ввести дозу как можно быстрее. Если до обычного времени введения следующей дозы осталось менее 12 ч, пациент не должен вводить пропущенную дозу, а должен возобновить введение препарата со следующей запланированной дозы. Не следует вводить дополнительную или повышенную дозу для компенсации пропущенной дозы.
Пациенты с СД2
Препарат Энлигрия® не следует применять в комбинации с другими агонистами рецепторов ГПП-1.
В начале терапии препаратом Энлигрия® рекомендуется уменьшить дозу одновременно применяемого препарата инсулина или секретагогов инсулина (таких как препараты сульфонилмочевины) для уменьшения риска развития гипогликемии. Самоконтроль концентрации глюкозы крови может быть необходим для корректировки дозы инсулина или секретагогов инсулина (см. раздел «Особые указания»).
Особые группы пациентов
Пациенты пожилого возраста (≥ 65 лет)
Коррекции дозы с учетом возраста не требуется. Опыт применения препарата у пациентов в возрасте ≥ 75 лет ограничен, применение препарата у этих пациентов не рекомендуется (см. подраздел «Фармакокинетика» и раздел «Противопоказания»).
Пациенты с почечной недостаточностью
У пациентов с почечной недостаточностью легкой или средней степени (КК ≥ 30 мл/мин) коррекции дозы не требуется. Применение препарата Энлигрия® у пациентов с почечной недостаточностью тяжелой степени (КК < 30 мл/мин), включая пациентов с терминальной стадией почечной недостаточности, противопоказано (см. подраздел «Фармакокинетика» и раздел «Противопоказания»).
Пациенты с печеночной недостаточностью
У пациентов с печеночной недостаточностью легкой и средней степени тяжести коррекции дозы не требуется. Применение препарата Энлигрия® у пациентов с печеночной недостаточностью тяжелой степени противопоказано (см. подраздел «Фармакокинетика» и раздел «Противопоказания»). У пациентов с печеночной недостаточностью легкой или средней степени тяжести препарат следует применять с осторожностью (см. подраздел «Фармакокинетика» и раздел «С осторожностью»).
Дети и подростки
У детей и подростков в возрасте от 12 лет и старше коррекции дозы не требуется.
Безопасность и эффективность препарата Энлигрия® у детей в возрасте до 12 лет не установлены (см. подраздел «Фармакодинамика»).
Способ применения
Препарат Энлигрия® предназначен только для подкожного введения. Его нельзя вводить внутривенно или внутримышечно.
Препарат Энлигрия® вводят один раз в сутки в любое время, независимо от приема пищи. Его следует вводить в область живота, бедра или плеча. Место и время инъекции могут быть изменены без коррекции дозы. Тем не менее, желательно делать инъекции примерно в одно и то же время суток после выбора наиболее удобного времени.
Резюме профиля безопасности
Безопасность лираглутида была оценена в 5 двойных слепых плацебо- контролируемых исследованиях, в которых приняли участие 5813 взрослых пациентов с ожирением или с избыточной массой тела и, как минимум, с одним связанным с избыточной массой тела сопутствующим заболеванием. В целом, нарушения со стороны ЖКТ являлись наиболее часто отмечаемыми нежелательными реакциями во время терапии лираглутидом (67,9 %) (см. подраздел «Описание отдельных нежелательных реакций»).
Табличное резюме нежелательных реакций
В Таблице 3 представлен перечень нежелательных реакций, зарегистрированных в ходе исследований для взрослых пациентов. Нежелательные реакции распределены по группам в соответствии с системами органов МеdDRА и частотой. Частота определена следующим образом: очень часто (≥ 1/10); часто (≥ 1/100 до < 1/10); нечасто (≥ 1/1 000 до < 1/100); редко (≥ 1/10 000 до < 1/1 000); очень редко (< 1/10 000); частота неизвестна (не может быть установлена по имеющимся данным).
Таблица 3. Нежелательные реакции, зарегистрированные в ходе исследований у взрослых пациентов
|
Очень часто |
Часто |
Нечасто |
Редко |
Частота неизвестна |
|
Нарушения со стороны иммунной системы |
||||
|
Анафилактические реакции |
||||
|
Нарушения метаболизма и питания |
||||
|
Гипогликемия* |
Дегидратация |
|||
|
Психические нарушения |
||||
|
Бессонница** |
||||
|
Нарушения со стороны нервной системы |
||||
|
Головная боль |
Головокружение |
|||
|
Дисгевзия |
||||
|
Нарушение со стороны сердца |
||||
|
Тахикардия |
||||
|
Желудочно-кишечные нарушения |
||||
|
Тошнота |
Сухость во рту |
Панкреатит*** |
||
|
Рвота |
Диспепсия |
Задержка опорожнения желудка**** |
||
|
Диарея |
Гастрит |
|||
|
Запор |
Гастроэзофагеаль-ная рефлюксная болезнь |
|||
|
Боль в верхних отделах живота |
||||
|
Метеоризм |
||||
|
Отрыжка |
||||
|
Вздутие живота |
||||
|
Нарушения со стороны печени и желчевыводящих путей |
||||
|
Холелитиаз*** |
Холецистит*** |
Холангит |
||
|
Холестатическая желтуха |
||||
|
Нарушения со стороны кожи и подкожных тканей |
||||
|
Крапивница |
||||
|
Нарушение со стороны почек и мочевыводящих путей |
||||
|
Острая почечная недостаточность |
||||
|
Нарушение функции почек |
||||
|
Общие нарушения и реакции в месте введения |
||||
|
Реакции в месте инъекции |
Недомогание |
|||
|
Астения |
||||
|
Утомляемость |
||||
|
Лабораторные и инструментальные данные |
||||
|
Повышение активности липазы |
||||
|
Повышение активности амилазы |
||||
* Гипогликемия (на основании отмеченных пациентами симптомов и не подтвержденная измерениями концентрации глюкозы крови), отмеченная у пациентов без СД2, получавших лираглутид в сочетании с диетой и физическими нагрузками. Подробную информацию см. в подразделе «Описание отдельных нежелательных реакций».
** Бессонницу отмечали во время первых 3-х месяцев терапии.
*** См. раздел «Особые указания».
**** В соответствии с фазами 2, 3а и 3b контролируемого клинического исследования.
Описание отдельных нежелательных реакций
Гипогликемия у пациентов без СД2
В КИ с участием пациентов с избыточной массой тела или ожирением без СД2, получавших терапию лираглутидом в сочетании с диетой и физическими нагрузками, тяжелых гипогликемий (требующих оказания помощи третьим лицом) отмечено не было. О симптомах гипогликемии сообщали 1,6 % пациентов, получавших лираглутид, и 1,1 % пациентов, получавших плацебо; однако эти случаи не были подтверждены измерениями концентрации глюкозы крови. В большинстве случаев отмечалась легкая гипогликемия.
Гипогликемия у пациентов с СД2
В КИ с участием пациентов с избыточной массой тела или ожирением и СД2, получавших терапию лираглутидом в сочетании с диетой и физическими нагрузками, случаи тяжелой гипогликемии (требующие оказания помощи третьим лицом) были отмечены у 0,7 % пациентов, получавших лираглутид, и только у пациентов, одновременно получавших терапию производными сульфонилмочевины. Также в этой группе пациентов подтвержденная гипогликемия была отмечена у 43,6 % пациентов, получавших лираглутид, и 27,3 % пациентов, получавших плацебо. Среди пациентов, не получавших одновременно препарат сульфонилмочевины, подтвержденная гипогликемия была отмечена у 15,7 % пациентов, получавших лираглутид, и у 7,6 % пациентов, получавших плацебо (концентрация глюкозы < 3,9 ммоль/л в сочетании с симптомами).
Гипогликемия у пациентов с СД2, получающих инсулин
В КИ с участием пациентов с избыточной массой тела или ожирением с СД2 типа, получавших терапию инсулином и лираглутидом в сочетании с диетой и физическими нагрузками и до двух пероральных гипогликемических препаратов, тяжелая гипогликемия (требующая оказания помощи третьим лицом) была отмечена у 1,5 % пациентов, получавших терапию лираглутидом. В данном исследовании подтвержденная гипогликемия (определяемая как уровень глюкозы в плазме ≤ 3,9 ммоль/л, сопровождаемая симптомами) была зарегистрирована у 47,2 % пациентов, получавших терапию лираглутидом, и у 51,8 % пациентов, получавших плацебо. Сообщалось о подтвержденных гипогликемических эпизодах среди пациентов, одновременно получавших производные сульфонилмочевины, у 60,9 % пациентов, получавших терапию лираглутидом, и у 60,0 % пациентов, получавших плацебо.
Нежелательные реакции со стороны желудочно-кишечного тракта
Большинство реакций со стороны ЖКТ были легкой или средней степени тяжести, преходящими и, в большинстве случаев, не приводили к прекращению терапии. Реакции обычно возникали в первые недели терапии, и их проявления постепенно уменьшались в течение нескольких дней или недель при продолжении терапии.
У пациентов в возрасте ≥ 65 лет могут наблюдаться более выраженные проявления нежелательных реакций со стороны ЖКТ во время терапии лираглутидом.
У пациентов с почечной недостаточностью легкой или средней степени тяжести (КК ≥ 30 мл/мин) могут наблюдаться более выраженные проявления нежелательных реакций со стороны ЖКТ во время терапии лираглутидом.
Острая почечная недостаточность
У пациентов на терапии агонистом рецепторов ГПП-1 отмечались случаи острой почечной недостаточности. Большинство зарегистрированных случаев произошло у пациентов, которые испытывали тошноту, рвоту или диарею, приводившую к уменьшению объема циркулирующей крови.
Аллергические реакции
В пострегистрационном периоде сообщалось о возникновении нескольких случаев анафилактических реакций с такими симптомами, как артериальная гипотензия, ощущение сердцебиения, одышка или периферические отеки. Анафилактические реакции потенциально могут быть угрожающими жизни. При подозрении на анафилактическую реакцию необходимо отменить терапию лираглутидом и не возобновлять лечение (см. раздел «Противопоказания»).
Реакции в месте введения
Сообщалось о реакциях в месте введения у пациентов, получавших лираглутид. Эти реакции, как правило, были легкой степени, носили транзиторный характер и в большинстве случаев исчезали при продолжении терапии.
Тахикардия
В КИ тахикардия была отмечена у 0,6 % пациентов, получавших лираглутид, и у 0,1 % пациентов, получавших плацебо. Большинство реакций были легкой или средней степени тяжести. Реакции были единичными и в большинстве случаев проходили при продолжении терапии лираглутидом.
Дети и подростки
В КИ, проведенном с участием подростков с ожирением в возрасте от 12 до 18 лет, 125 пациентов получали терапию лираглутидом в течение 56 недель.
В целом, частота развития, тип и степень тяжести нежелательных реакций у подростков с ожирением были сопоставимы с таковыми у взрослых пациентов. Рвота у подростков возникала в два раза чаще по сравнению со взрослыми пациентами.
Количество пациентов, сообщивших хотя бы об одном эпизоде клинически значимой гипогликемии, было выше в группе терапии лираглутидом (1,6 %) по сравнению с плацебо (0,8 %). Эпизодов тяжелой гипогликемии в ходе исследования выявлено не было.
По данным КИ и пострегистрационного применения лираглутида были зарегистрированы случаи передозировки при применении препарата в дозе до 72 мг (в 24 раза больше рекомендуемой дозы для коррекции массы тела).
Пациенты отмечали сильную тошноту, сильную рвоту и тяжелую гипогликемию.
В случае передозировки необходимо начать соответствующую поддерживающую терапию в соответствии с клиническими признаками и симптомами.
Пациента следует наблюдать на предмет клинических признаков дегидратации и контролировать концентрацию глюкозы крови.
In vitro лираглутид показал очень низкую способность к лекарственному фармакокинетическому взаимодействию, обусловленному метаболизмом в системе цитохрома Р-450 (CYP), а также связыванием с белками плазмы.
Небольшая задержка опорожнения желудка при применении лираглутида может влиять на всасывание одновременно применяемых препаратов для приема внутрь. Исследования лекарственного взаимодействия не показали какого-либо клинически значимого замедления всасывания этих препаратов, поэтому коррекции дозы не требуется.
Исследования взаимодействия проводили с применением лираглутида в дозе 1,8 мг. Влияние на скорость опорожнения желудка было одинаковым при применении лираглутида в дозе 1,8 мг и 3,0 мг (AUC0-300 мин парацетамола). У нескольких пациентов, получавших лечение лираглутидом, отмечалось как минимум по одному эпизоду тяжелой диареи. Диарея может оказывать влияние на всасывание пероральных лекарственных препаратов, которые применяются одновременно с лираглутидом.
Варфарин и другие производные кумарина
Исследования по взаимодействию не проводились. Не может быть исключено клинически значимое взаимодействие с действующими веществами с низкой растворимостью или узким терапевтическим индексом, такими как варфарин. В начале лечения лираглутидом у пациентов, получающих варфарин или другие производные кумарина, рекомендуется чаще проводить мониторинг международного нормализованного отношения (МНО).
Парацетамол (ацетаминофен)
Лираглутид не изменял общую экспозицию парацетамола после введения однократной дозы парацетамола 1000 мг. Максимальная концентрация (Сmах) парацетамола была снижена на 31 %, а медиана tmax увеличена на 15 мин. Коррекции дозы при сопутствующем применении парацетамола не требуется.
Аторвастатин
Лираглутид не изменял общую экспозицию аторвастатина после применения однократной дозы аторвастатина 40 мг. Поэтому коррекции дозы аторвастатина при применении в сочетании с лираглутидом не требуется. Сmах аторвастатина была снижена на 38 %, а медиана tmax увеличена с 1 ч до 3 ч при применении лираглутида.
Гризеофульвин
Лираглутид не изменял общую экспозицию гризеофульвина после применения однократной дозы гризеофульвина 500 мг. Сmах гризеофульвина была увеличена на 37 %, а медиана tmax не изменилась. Коррекции дозы гризеофульвина и других соединений с низкой растворимостью и высокой проникающей способностью не требуется.
Дигоксин
Применение однократной дозы дигоксина 1 мг в сочетании с лираглутидом привело к уменьшению AUC дигоксина на 16 %, уменьшению Сmах на 31 %. Медиана tmax дигоксина увеличилась с 1 ч до 1,5 ч. С учетом данных результатов коррекции дозы дигоксина не требуется.
Лизиноприл
Применение однократной дозы лизиноприла 20 мг в сочетании с лираглутидом привело к уменьшению AUC лизиноприла на 15 %, уменьшению Сmах на 27 %. При применении лираглутида медиана tmax лизиноприла увеличилась с 6 ч до 8 ч. С учетом данных результатов коррекции дозы лизиноприла не требуется.
Пероральные контрацептивы
Лираглутид приводил к уменьшению Сmах этинилэстрадиола и левоноргестрела на 12 % и 13 %, соответственно, после применения однократной дозы перорального гормонального контрацептивного препарата. tmах обоих лекарственных веществ на фоне применения лираглутида увеличивалась на 1,5 ч. Не было отмечено клинически значимого влияния на системную экспозицию этинилэстрадиола или левоноргестрела. Таким образом, не ожидается влияния на контрацептивный эффект при совместном применении с лираглутидом.
Дети и подростки
Исследования взаимодействия проводились только на взрослых пациентах.
Несовместимость
Лекарственные вещества, добавленные к лираглутиду, могут вызвать разрушение лираглутида. В связи с отсутствием исследований совместимости данный лекарственный препарат нельзя смешивать с другими лекарственными препаратами.
Панкреатит
Применение агонистов рецепторов ГПП-1 ассоциировалось с развитием острого панкреатита.
Пациенты должны быть проинформированы о характерных симптомах острого панкреатита. В случае подозрения на развитие панкреатита применение препарата Энлигрия® следует прекратить; в случае подтверждения острого панкреатита терапию препаратом Энлигрия® возобновлять не следует.
Холелитиаз и холецистит
В КИ была отмечена более высокая частота развития холелитиаза и холецистита у пациентов, получавших лираглутид, по сравнению с получавшими плацебо пациентами. Это может быть частично объяснено тем, что значительное снижение массы тела при применении лираглутида может увеличить риск развития холелитиаза и, следовательно, холецистита. Холелитиаз и холецистит могут привести к госпитализации и холецистэктомии.
Пациенты должны быть проинформированы о характерных симптомах холелитиаза и холецистита.
Заболевания щитовидной железы
В ходе КИ с участием пациентов с СД2 были отмечены нежелательные реакции со стороны щитовидной железы, включая увеличение концентрации кальцитонина в сыворотке крови, зоб и новообразование щитовидной железы, в особенности у пациентов, уже имеющих заболевания щитовидной железы. У пациентов с заболеваниями щитовидной железы лираглутид следует применять с осторожностью.
В постмаркетинговом периоде у пациентов, получавших лираглутид, были отмечены случаи медуллярного рака щитовидной железы. Имеющихся данных недостаточно для установления или исключения причинно-следственной связи возникновения медуллярного рака щитовидной железы с применением лираглутида у человека. Препарат Энлигрия® противопоказан к применению у пациентов с медуллярным раком щитовидной железы в анамнезе, в том числе в семейном, и множественной эндокринной неоплазией 2 типа. Необходимо проинформировать пациента о риске медуллярного рака щитовидной железы и о симптомах опухоли щитовидной железы (уплотнения в области шеи, дисфагия, одышка, непроходящая охриплость голоса).
Текущий контроль концентрации кальцитонина в сыворотке крови или ультразвуковое исследование (УЗИ) щитовидной железы не имеют существенного значения для раннего выявления медуллярного рака щитовидной железы у пациентов, применяющих лираглутид. Значительное повышение концентрации кальцитонина в сыворотке крови может свидетельствовать о наличии медуллярного рака щитовидной железы, пациенты с медуллярным раком щитовидной железы обычно имеют концентрацию кальцитонина более 50 нг/л. При выявлении повышения концентрации кальцитонина в сыворотке крови необходимо провести дальнейшее обследование пациента. Пациенты с узлами щитовидной железы, выявленными при медосмотре или при УЗИ щитовидной железы, также должны быть дополнительно обследованы.
Частота сердечных сокращений
В КИ было отмечено увеличение ЧСС (см. подраздел «Клиническая эффективность и безопасность»). Следует проводить контроль ЧСС с интервалами, соответствующими обычной клинической практике. Пациентов следует проинформировать о симптомах тахикардии (ощущение сердцебиения или ощущение учащенного сердцебиения в покое). У пациентов с клинически значимой постоянной тахикардией в состоянии покоя следует прекратить терапию препаратом Энлигрия®.
Дегидратация
Признаки и симптомы дегидратации, включая нарушение функции почек и острую почечную недостаточность, были отмечены у пациентов, получавших агонисты рецепторов ГПП-1.
Пациенты, получающие препарат Энлигрия®, должны быть проинформированы о потенциальном риске дегидратации, связанном с нежелательными реакциями со стороны ЖКТ, и о необходимости профилактики гиповолемии.
Гипогликемия у пациентов с СД2
Риск развития гипогликемии может быть выше у пациентов с СД2, получающих лираглутид в комбинации с инсулином и/или производными сульфонилмочевины. Этот риск может быть уменьшен путем снижения дозы инсулина и/или производного сульфонилмочевины.
Дети и подростки
Сообщалось об эпизодах клинически значимой гипогликемии у подростков (≥ 12 лет), получавших терапию лираглутидом. Пациенты должны быть проинформированы о характерных симптомах гипогликемии и соответствующих действиях.
Гипергликемия у пациентов с сахарным диабетом, получавших инсулин
У пациентов с СД препарат Энлигрия® не должен применяться в качестве замены инсулина.
Сообщалось о диабетическом кетоацидозе у инсулинзависимых пациентов после быстрой отмены или снижения дозы инсулина (см. раздел «Способ применения и дозы»).
Вспомогательные вещества
Препарат Энлигрия® содержит менее 1 ммоль (23 мг)/доза натрия, то есть, по сути, не содержит натрия.
Суицидальные мысли и поведение
В ходе КИ 6 (0,2 %) из 3384 пациентов, получавших лираглутид, сообщили о появлении суицидальных мыслей, один из пациентов предпринял попытку суицида. У пациентов (1941 человек), получавших плацебо, это отмечено не было. Пациентов необходимо контролировать в отношении появления или ухудшения депрессии, суицидальных мыслей или поведения, и/или любых неожиданных изменений в настроении или поведении. У пациентов с суицидальными мыслями или поведением применение препарата Энлигрия® следует прекратить.
Противопоказано применять препарат Энлигрия® у пациентов с суицидальными попытками или активными суицидальными мыслями в анамнезе.
Рак молочной железы (РМЖ)
В ходе КИ сообщалось о подтвержденном РМЖ у 14 (0,6 %) из 2379 женщин, получавших лираглутид, по сравнению с 3 (0,2 %) из 1300 женщин, получавших плацебо, включая инвазивный рак (11 случаев у женщин, получавших лираглутид, и 3 случая у женщин, получавших плацебо) и внутрипротоковую карциному in situ (3 случая у женщин, получавших лираглутид, и 1 случай у женщины, получавшей плацебо). Большинство случаев рака были эстроген- и прогестерон зависимыми. Невозможно определить, были ли эти случаи связаны с применением лираглутида из-за их слишком небольшого количества.
Кроме того, нет достаточных данных, чтобы определить, оказывает ли лираглутид влияние на уже существующие новообразования молочной железы.
Папиллярный рак щитовидной железы
В ходе КИ сообщалось о подтвержденной папиллярной карциноме щитовидной железы у 7 (0,2 %) из 3291 пациентов, получавших лираглутид, по сравнению с отсутствием ее в группе пациентов, получавших плацебо (1843 пациента). Из всех случаев 4 карциномы были менее 1 см в наибольшем диаметре и 4 были диагностированы по результатам гистологии после проведенной по медицинским показаниям тиреоидэктомии.
Неоплазии ободочной и прямой кишки
В ходе КИ сообщалось о подтвержденных доброкачественных неоплазиях ободочной и прямой кишки (преимущественно аденомах ободочной кишки) у 17 (0,5 %) из 3291 пациентов, получавших лираглутид, по сравнению с 4 (0,2 %) из 1843 пациентов, получавших плацебо. Было зарегистрировано два подтвержденных случая злокачественной карциномы ободочной и прямой кишки (0,1 %) у пациентов, получавших лираглутид, и ни одного у пациентов, получавших плацебо.
Нарушения сердечной проводимости
В ходе КИ у 11 (0,3 %) из 3384 пациентов, получавших лираглутид, сообщалось о развитии нарушений сердечной проводимости, таких как атриовентрикулярная блокада 1 степени, блокада правой ножки пучка Гиса или блокада левой ножки пучка Гиса. У пациентов (1941 человек), получавших плацебо, о развитии нарушений сердечной проводимости не сообщалось.
Лираглутид не влияет или незначительно влияет на способность управлять транспортными средствами и механизмами. Однако существует риск возникновения головокружения, в основном в течение первых 3 месяцев лечения препаратом Энлигрия®. При возникновении головокружения следует соблюдать осторожность при управлении транспортным средством или работе с механизмами.
В связи с риском развития гипогликемии при применении препарата, особенно при комбинированном применении с препаратами сульфонилмочевины у пациентов с СД2, следует соблюдать осторожность при управлении транспортными средствами и механизмами.
Препарат Энлигрия® нельзя применять, если он выглядит иначе, чем прозрачная бесцветная или коричневого цвета жидкость.
Препарат Энлигрия® нельзя применять, если он подвергся замораживанию.
Шприц-ручка предназначена для введения препарата Энлигрия® в картриджах 3,0 мл. Совместима с любыми иглами для шприц-ручек.
Пациент должен быть проинформирован о том, что использованную иглу следует выбрасывать после каждой инъекции, а также о том, что нельзя хранить шприц-ручку с присоединенной иглой. Такая мера позволит предотвратить загрязнение, инфицирование и утечку препарата из шприц-ручки и гарантирует точность дозирования.
Шприц-ручка предназначена только для индивидуального использования.
Весь оставшийся лекарственный препарат и отходы следует уничтожить в соответствии с установленными национальным законодательством требованиями.
Раствор для подкожного введения, 6 мг/мл.
По 3 мл препарата в картриджи из бесцветного стекла 1-го гидролитического класса вместимостью 3 мл, укупоренные плунжерами из бромбутиловой резины с одной стороны и дисками из бромбутиловой резины/полиизопрена, обкатанные алюминиевыми колпачками, с другой стороны.
Картриджи устанавливают в пластиковую мультидозовую одноразовую шприц-ручку для многократных инъекций.
Каждая пластиковая мультидозовая одноразовая шприц-ручка для многократных инъекций градуирована для возможности установки следующих вариантов доз на одно введение: 0,6 мг – 1,2 мг – 1,8 мг – 2,4 мг –3,0 мг (шаг: 0,6 мг).
На каждую шприц-ручку наклеивают самоклеящуюся этикетку.
По 1, 2, 3, 4, 5 или 6 пластиковых мультидозовых одноразовых шприц-ручек для многократных инъекций в контурной ячейковой упаковке или без контурной ячейковой упаковки вместе с инструкцией по медицинскому применению помещают в картонную пачку с контролем первого вскрытия или без него.
Дополнительно пачка с пластиковыми мультидозовыми одноразовыми шприц-ручками для многократных инъекций может комплектоваться стерильными иглами для мультидозовой одноразовой шприц-ручки для одноразового использования в количестве 10, 15, 20, 25, 30 штук в контурной ячейковой упаковке или без контурной ячейковой упаковки.
Хранить в недоступном для детей месте.
Хранить при температуре от 2 до 8 °С. Хранить в оригинальной упаковке (пачке). Используемую шприц-ручку с препаратом хранить при температуре не выше 30 °С или в холодильнике (при температуре от 2 до 8 °С). Использовать в течение 1 месяца. Закрывать шприц-ручку колпачком для защиты от света.
Препарат не замораживать.
2,5 года.
Не применять по истечении срока, указанного на этикетке шприц-ручки и упаковке.
Отпускают по рецепту.
ООО «ПРОМОМЕД РУС», Россия
129090, г. Москва, проспект Мира, дом 13, строение 1, офис 13
АО «Биохимик», Россия
Юридический адрес:
430030, Республика Мордовия, г. Саранск, ул. Васенко, д. 15А
Адрес места производства:
430030, Республика Мордовия, г. Саранск, ул. Васенко, д. 15А
Телефон: +7 (8342) 38-03-68
Адрес электронной почты: biohimic@promomed.pro
Адрес в сети в интернет: www.promomed.ru
ООО «Завод Медсинтез», Россия
Юридический адрес:
620028, Свердловская обл., г. Екатеринбург, ул. Кирова, стр. 28, пом. 205
Адрес места производства:
624130, Свердловская обл., г. Новоуральск, ул. Торговая, зд. 15, стр. 3
Телефон: +7 (34370) 2-50-61
Факс: +7 (34370) 2-54-95
ООО «ПРОМОМЕД РУС», Россия
129090, г. Москва, проспект Мира, дом 13, строение 1, офис 13
Телефон: +7 (495) 640-25-28
Круглосуточный телефон горячей линии фармаконадзора: 8-800-777-86-04 (бесплатно)
Адрес электронной почты: reception@promomed.pro
Вся информация, размещенная в данном веб-сайте, предназначена исключительно для специалистов здравоохранения/медицинских работников.
Пожалуйста, подтвердите, что Вы являетесь медицинским или фармацевтическим работником.







